|
Show all abstracts Show selected abstracts Add to my list |
|
| EDITORIALS |
|
|
|
Annoucement |
p. 271 |
Kanjaksha Ghosh
DOI:10.4103/0971-6866.107970 PMID:23716930 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Small players with a big role: MicroRNAs in pathophysiology of cleft lip and palate |
p. 272 |
Uppala Radhakrishna
DOI:10.4103/0971-6866.107973 PMID:23716931 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Vitamin D receptor and type 2 diabetes mellitus: Growing therapeutic opportunities |
p. 274 |
Parmeet Kaur Manchanda, Hemant K Bid
DOI:10.4103/0971-6866.107975 PMID:23716932 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| REVIEW ARTICLE |
 |
|
|
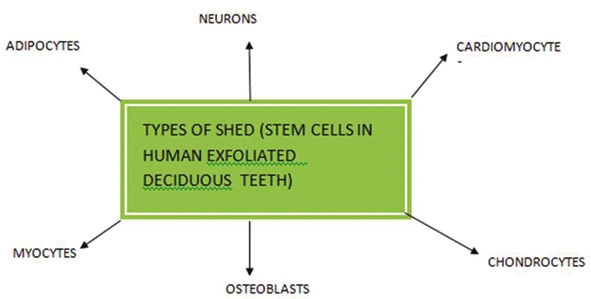  |
Redefining the potential applications of dental stem cells: An asset for future |
p. 276 |
Shalu Rai, Mandeep Kaur, Sandeep Kaur, Sapna Panjwani Arora
DOI:10.4103/0971-6866.107976 PMID:23716933Recent exciting discoveries isolated dental stem cells from the pulp of the primary and permanent teeth, from the periodontal ligament, and from associated healthy tissues. Dental pulp stem cells (DPSCs) represent a kind of adult cell colony which has the potent capacity of self-renewing and multilineage differentiation. Stem cell-based tooth engineering is deemed as a promising approach to the making of a biological tooth (bio-tooth) or engineering of functional tooth structures. Dental professionals have the opportunity to make their patients aware of these new sources of stem cells that can be stored for future use as new therapies are developed for a range of diseases and injuries. The aim of this article is to review and understand how dental stem cells are being used for regeneration of oral and conversely nonoral tissues. A brief review on banking is also done for storing of these valuable stem cells for future use. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| ORIGINAL ARTICLES |
|
|
|
|
Evaluation of C677T polymorphism of the methylenetetra hydrofolate reductase gene and its association with levels of serum homocysteine, folate, and vitamin B12 as maternal risk factors for Down syndrome |
p. 285 |
Pankaj K Mohanty, Seema Kapoor, Anand P Dubey, Sanjeev Pandey, Renuka Shah, Hemant K Nayak, Sunil K Polipalli
DOI:10.4103/0971-6866.107977 PMID:23716934Aims and Objective: Evaluation of C677T polymorphisms of the methylenetetra hydrofolate reductase (MTHFR) gene and its association with level of serum homocysteine, folate, and vitamin B12 as possible maternal risk factors for Down syndrome.
Design: This was a case-control study.
Material and Methods : Fifty-two mothers (mean age 27.6 years) with babies having free trisomy 21 of North Indian ethnicity and 52 control nonlactating mothers (mean age 24.9 years) of same ethnicity attending services of genetic lab for bloodletting for other causes were enrolled after informed written consent. Fasting blood was collected and was used for determination of plasma homocysteine, vitamin B12, and folate (serum and RBC), and for PCR amplification of the MTHFR gene.
Results: The prevalence of MTHFR C677T polymorphism in north Indian mothers of babies with trisomy 21 Down syndrome was 15.38% compared to 5.88 % in controls. The difference between two groups was not statistically significant ( P = 0.124). Low serum folate was demonstrated in 34.62% of cases vs. 11.54% in controls, which was significant ( P = 0.005). Low RBC folate was found in 30.7% of cases versus 11.53% in controls, which was not significant ( P = 0.059), when analyzed independently. But on multiple regression analysis the difference was statistically significant. Low serum vitamin B12 was found in 42.31% of cases versus 34.62% in controls, which was not significant ( P = 0.118). The mean serum homocysteine in cases was 10.35 ± 0.68 while controls were 9.02 ± 0.535.
Conclusion: Serum levels of folate were low in cases. The RBC folate levels were comparable in both groups. However the combined serum folate and RBC folate were low in cases compared to control groups. Homocysteine levels in our study were higher in Down syndrome mothers compared to controls; however high-serum level of Homocysteine had no association with MTHFR polymorphism. No association of serum vitamin B12 with MTHFR polymorphism in occurrence of Down syndrome births was found. Peri- or preconceptional folate supplementation may therefore lead to a decline in DS births, if supported by larger studies. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Molecular analysis of exons 6 and 7 of phenylalanine hydroxylase gene mutations in Phenylketonuria patients in Western Iran |
p. 290 |
Keyvan Moradi, Reza Alibakhshi, Keyghobad Ghadiri, Saeid Reza Khatami, Hamid Galehdari
DOI:10.4103/0971-6866.107978 PMID:23716935Background: Phenylketonuria (PKU) is an inborn error of amino acid metabolism that results from a deficiency of phenylalanine hydroxylase (PAH). According to PAH database, exons 6 and 7 and their flanking introns of PAH gene contain the greatest number of mutant alleles. Therefore, as a preliminary study, nucleotide sequence analysis of exons 6 and 7 of the PAH gene has been performed in 25 PKU patients whose ancestors lived in Kermanshah province of Iran. To date, there has been no mutation data describing the genotypes of the PKU disease in this Kurdish ethnic region background.
Materials and Methods: Twenty-five patients (aged between 2 and 23 years) participated in this study. The DNA fragments containing two exons of the PAH gene [6 and 7] and their exon-flanking intronic sequences were amplified and sequenced.
Results: The total of detected mutations were R261X (8%), R176X (4%), R243Q (4%), R243X (2%) and R261Q (2%), as they accounted for 20% of all mutant alleles in this study. The identified polymorphisms are: IVS5 -54 G > A (22%), Q232Q (8%) and V245V (4%). All of the detected mutations in this study are related to CpG dinucleotides in the PAH gene sequence.
Conclusion: The frequency of R261X, the most common mutation in our study, in Iranian population is <5%. Furthermore, there is no report of detection of R176X and R243Q in Isfahan and Azeri Turkish populations. These findings confirm the common Mediterranean mutations in this local population, although with more or lower frequencies than those reported in other related studies in Iran. Therefore, it may be necessary to study the PAH gene mutations in other provinces of Iran separately. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Hemoglobinopathies in South Gujarat population and incidence of anemia in them |
p. 294 |
Ankur G Patel, Avani P Shah, Smita M Sorathiya, Snehalata C Gupte
DOI:10.4103/0971-6866.107979 PMID:23716936Objective: To Screen of South Gujarat population for determination of prevalence of different hemoglobinopathies particularly beta thalassemia trait (BTT) and sickle cell trait (SCT) and find out the incidence of anemia in them.
Material and Methods: The present study screened 32,857 samples of students from different school and colleges in South Gujarat. Blood samples were initially tested for solubility test and complete hemogram on hematology analyzer. Samples having MCV (≤78), MCH (≤28) and/or positive solubility test were investigated for Hb electrophoresis on cellulose acetate membrane (pH 8.6). Hb A 2 level ≥3.5% was considered as diagnostic for BTT. High performance liquid chromatography on Biorad Hb variant system was done on samples having doubtful results.
Result: Overall prevalence of BTT and SCT in South Gujarat was 4.4% and 1.3% respectively. Gamit, Vasava, Chaudhary, and Mahyavanshi castes had high prevalence of BTT (15.9%, 13.6%, 12.6%, and 6.9%) as well as SCT (22.2%, 15.2, 22.3, and 4.2%) respectively. Other communities like Lohana (10.8%), Sindhi (10.2%), Prajapati (6.3%), and Ghanchi (6.2%) also showed higher prevalence of BTT. Incidence of mild to moderate anemia was higher in BTT and SCT compared to non-BTT or non-SCT subjects.
Conclusion : Study suggests that BTT is the most prevalent hemoglobinopathy in South Gujarat. β-thalassemia and Sickle cell anemia are highly prevalent in Mahyavanshi, Chaudhary, Gamit, Vasava and Rohit. Prajapati, Lohana, Leva Patel, and Ghanchi have β- thalassemia risk. SCT is more frequently detected in Dhodia Patel and Kukanas. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
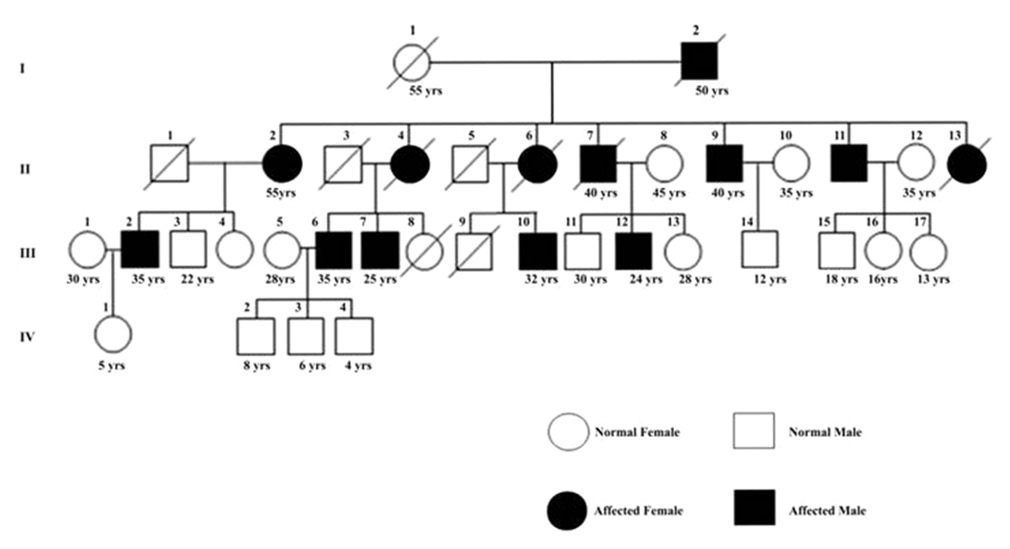 |
Analysis of autosomal dominant spinocerebellar ataxia type 1 in an extended family of central India |
p. 299 |
Shashikant Sharma, Tekcham Dinesh Singh, Satish S Poojary, Manoj Singh Rohilla, Ajaypal Singh, Kishore B Lowalekar, Pramod Kumar Tiwari
DOI:10.4103/0971-6866.107981 PMID:23716937Background: Spinocerebeller ataxia type 1 (SCA1) is a specific type of ataxia among a group of inherited diseases of the central nervous system. In SCA1, genetic defects lead to impairment of specific nerve fibers carrying messages to and from the brain, resulting in the degeneration of the cerebellum, the coordination center of the brain. We investigated 24 members of an extended family in Gwalior city, India, some of which were earlier clinically diagnosed to be suffering from yet unconfirmed type of SCA neurodegenerative disorder.
Materials and Methods: All the family members from each age group were screened clinically and the characteristics of those resembling with ataxia were recorded for diagnosis by MRI. The confirmed patients of the family were genetically tested by PCR based molecular testing to identify the type of SCA (i.e., SCA 1, 2, 3, 4, 6 or 7). Family tree of the disease inheritance was constructed by pedigree based method.
Result and Conclusion: We found the clinical (symptoms and MRI) and genetic (Pedigree and PCR) results to be correlated. The PCR result revealed the disease to be of SCA 1 type being inherited in the family. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
The use of hormones indicators in human saliva in diagnosing parodontitis in pregnant women |
p. 305 |
SI Dolomatov, W Zukow, ID Atmazhov, R Muszkieta, A Skaliy
DOI:10.4103/0971-6866.107982 PMID:23716938Aims: The purpose of this work- was to study the dynamics of biochemical parameters of human saliva and analyze the features of the chemical composition of the saliva of women with abnormal pregnancy and in periodontitis against pregnancy.
Materials and Methods: The study included four groups of women: a control group of nonpregnant women of childbearing age (10), pregnant women with physiological pregnancy (24-28 weeks) without any signs of periodontal disease (10), pregnant with a generalized periodontitis I--II degrees in remission (10), women with pathological pregnancy with no signs of periodontal inflammation (10). In each of the groups over two samples of saliva were collected, the first collection of saliva in the morning on an empty stomach. Then mouthwash 0.9% sodium chloride solution was assigned and after 30 minutes the second portion of saliva. By enzyme immunoassay in samples of saliva of control groups of nonpregnant and pregnant women, as well as women with signs of a pathological course of pregnancy, the content of estriol, testosterone, and dehydroepiandrosterone sulfate was determined.
Statistical Analysis Used: Statistical data analysis was performed by the standard technique using Student's t-test.
Results: The results of biochemical analysis of saliva samples collected before rinsing the mouth with saline in groups of healthy nonpregnant and pregnant women were compared. It was established that during pregnancy the concentration of salivary estriol increases, but in pregnant women with periodontitis, the amount of this hormone in the saliva was significantly reduced. The highest content of testosterone in saliva samples, observed in healthy pregnant women, was significantly higher than nonpregnant women.
In pregnant women with periodontitis concentration of testosterone in saliva is reduced, while remaining significantly higher than its level in the saliva of nonpregnant women.
The highest concentration of testosterone is observed in the saliva of healthy pregnant women with periodontitis, but the smallest concentration of testosterone is found in the saliva of nonpregnant women. Also the nonpregnant group has the lowest levels of DHEA in pregnancy, and its content increases almost threefold when periodontal disease further grows.
Conclusions: It was established that periodontitis against pregnancy is characterized by higher levels of salivary DHEA sulfate and lower estriol, compared with a control group of pregnant women. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
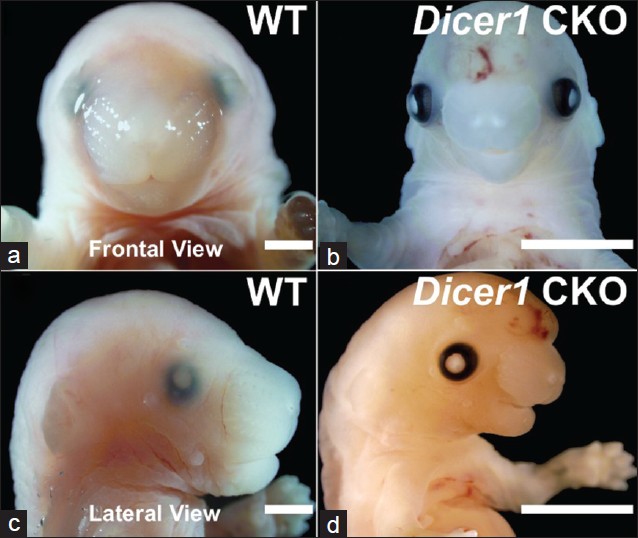 |
Conditional deletion of the human ortholog gene Dicer1 in Pax2-Cre expression domain impairs orofacial development |
p. 310 |
Laura C Barritt, Joseph M Miller, Laura R Scheetz, Kelsey Gardner, Marsha L Pierce, Garrett A Soukup, Sonia M Rocha-Sanchez
DOI:10.4103/0971-6866.107984 PMID:23716939Background: Orofacial clefts are common worldwide and result from insufficient growth and/or fusion during the genesis of the derivatives of the first pharyngeal arch and the frontonasal prominence. Recent studies in mice carrying conditional and tissue-specific deletions of the human ortholog Dicer1, an RNAse III family member, have highlighted its importance in cell survival, differentiation, proliferation, and morphogenesis. Nevertheless, information regarding Dicer1 and its dependent microRNAs (miRNAs) in mammalian palatogenesis and orofacial development is limited.
Aims: To describe the craniofacial phenotype, gain insight into potential mechanisms underlying the orofacial defects in the Pax2-Cre/Dicer1 CKO mouse, and shed light on the role of Dicer1 in mammalian palatogenesis.
Materials And Methods: Histological and molecular assays of wild type (WT) and Pax2-Cre/Dicer1 loxP/loxP (Dicer1 CKO) mice dissected tissues have been performed to characterize and analyze the orofacial dysmorphism in Pax2-Cre/Dicer1 loxP/loxP mouse.
Results: Dicer1 CKO mice exhibit late embryonic lethality and severe craniofacial dysmorphism, including a secondary palatal cleft. Further analysis suggest that Dicer1 deletion neither impacts primary palatal development nor the initial stages of secondary palatal formation. Instead, Dicer1 is implicated in growth, differentiation, mineralization, and survival of cells in the lateral palatal shelves. Histological and molecular analysis demonstrates that secondary palatal development becomes morphologically arrested prior to mineralization around E13.5 with a significant increase in the expression levels of apoptotic markers (P < 0.01).
Conclusions: Pax2-Cre-mediated Dicer1 deletion disrupts lateral palatal outgrowth and bone mineralization during palatal shelf development, therefore providing a mammalian model for investigating the role of miRNA-mediated signaling pathways during palatogenesis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
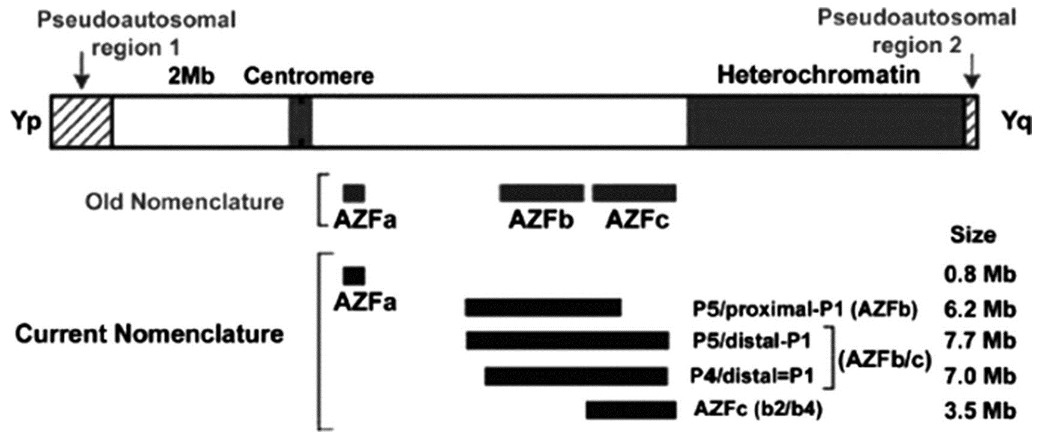 |
The low frequency of Y chromosome microdeletions in subfertile males in a Sinhalese population of Sri Lanka |
p. 320 |
Tithila Kalum Wettasinghe, Rohan W Jayasekara, Vajira H. W. Dissanayake
DOI:10.4103/0971-6866.107985 PMID:23716940Aims: This study was designed to determine the prevalence of azoospermia factor (AZF) microdeletions on the Y chromosome in Sri Lankan Sinhalese infertile men with azoospermia and severe oligozoospermia.
Settings and Design: The patient group was 207 karyotypically normal infertile Sinhalese males.
Materials and Methods: The presence of 13 sequence-tagged site (STS) markers in the AZF region was tested using multiplex polymerase chain reaction (M-PCR). One hundred and twenty unselected men were also studied as a control group.
Results: Three (1.5%) had classic Y chromosome microdeletions in the AZFc sub-region.
Conclusions: These results suggest a much lower Y chromosome microdeletion frequency than previously thought, even among a strictly selected group of sub-fertile males in Sri Lanka. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
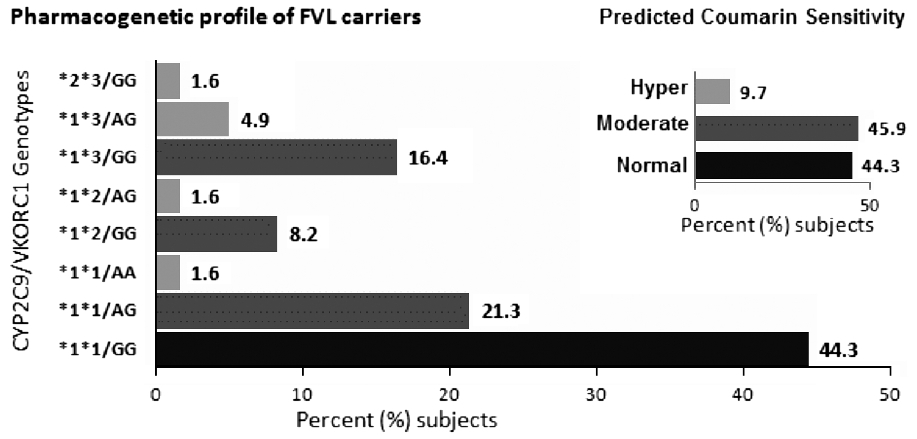 |
Pharmacogenetic typing for oral anti-coagulant response among factor V Leiden mutation carriers |
p. 326 |
Risha Nahar, Renu Saxena, Roumi Deb, Ishwar C Verma
DOI:10.4103/0971-6866.107987 PMID:23716941Context: Factor V Leiden mutation is the most common inherited predisposition for hypercoagulability and thereby a common genetic cause for initiation of oral anti-coagulation therapy. There is a dearth of knowledge of coumarin response profile in such thrombophilic population.
Aims: The current pilot study aims to estimate coumarin sensitivity in an Indian cohort with an inherited thrombophilia risk factor (Factor V Leiden mutation carriers) based on the observed frequency of CYP2C9 *2, *3 and VKORC1-1639G >A genotype combinations.
Settings and Design: A retrospective study carried out in a tertiary health care center in India.
Materials and Methods: Carriers of FVL mutation were genotyped for CYP2C9 (*2, *3) and VKORC1 (-1639G >A) variants by PCR-RFLP technique.
Statistical Analysis Used: Chi-square test to analyze difference in expected and observed genotype frequency.
Results: Sixty-one (n = 61) unrelated carriers of FVL mutation were observed in the 13 years study period. The allele frequency of CYP2C9 *2, CYP2C9 *3, and VKORC1-1639A in this cohort was 0.06, 0.11, and 0.16, respectively. Six (9.7%) individuals had two of the three variant alleles (heterozygous or homozygous), and 28 (45.9%) were heterozygous for at least one polymorphism.
Conclusions: Pre-prescription genotyping for coumarin drugs, if introduced in Indians with inherited thrombophilia (in whom oral anti-coagulant therapy may be necessary), is likely to identify 9.7% (hypersensitive) subjects in whom the optimum anti-coagulation may be achieved with reduced dosages, 44.3% (normal sensitivity) who may require higher dose and also 55.6% (hyper and moderate sensitivity) subjects who are likely to experience bleeding episodes. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Genetic significance of muscle segment homeo box1 gene in South Indian population for cleft lip and palate |
p. 332 |
S Prasad Venkanna, Venkatesh Shivani
DOI:10.4103/0971-6866.107988 PMID:23716942Background: Oral clefts having a prehistoric existence and the latest scientific technologies have shown new insights in identifying the cause and management. So this is a DNA/gene based study has been presented in this article which comprises the significance of MSX1 gene in cleft samples of major states of South India.
Aims: To evaluate the significance of MSX1 gene in South Indian population having cleft lip and palate.
Settings and Design: Four states of native population were set for the study. From each state renowned cleft operating center was selected with the prior ethical and suitable permission and patient consent was taken. Blood samples were collected from each effected sibling were studied and their details were coded. The collected blood samples were used for DNA isolation, PCR amplification and sequencing.
Materials and Methods: Eighty patients with non-syndromic CL/CLP/CP from various cleft operating centers in southern states (Karnataka, Tamilnadu, Kerala and Andhra Pradesh) with different ethnic/cultural background were taken. Twenty samples (families) were collected from each state and sequenced and compared with earlier data.
Results: Analysis of this study indicates that mutation of either G273A/C or C102G seems to cause cleft formation. In this analysis, we found a novel mutation (414G to T) which is submitted to NCBI Gene data bank (EF065625).
Conclusion: This study supports MSX1 gene leading to cleft lip and palate in the samples studied. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Analysis of loss of heterozygsity effect on thyroid tumor with oxyphilia cell locus in familial non medullary thyroid carcinoma in Iranian families |
p. 340 |
Hasti Atashi Shirazi, Mehdi Hedayati, Maryam Sadat Daneshpour, Abdollah Shafiee, Fereidoun Azizi
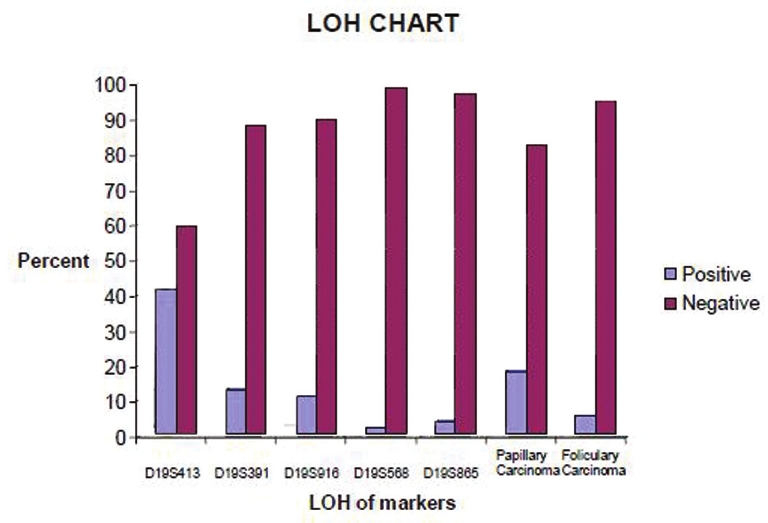
DOI:10.4103/0971-6866.107989 PMID:23716943Material and Methods: 22 nuclear families (78 persons including 12 patients) with papillary and follicular tumors were selected in a period of six months from Milad hospital. Five microsatellite markers (D19S413, D19S391, D19S916, D19S568, D19S865) on 19p13.2 were selected for genetic analysis. Genomic DNAs was extracted; PCR and polyacrylamide gel electrophoresis method were used for variation detection.
Results: The results show that 5.4% of the follicular carcinomas and 17.9% of the papillary carcinomas presented LOH at recognition sites. LOH of Papillary carcinoma detected about 13.9% and follicular carcinoma 7.2% in this study. The frequency of informative cases was not similar for each marker: D19S413 (41.1%)[1], D19S391 (12.5%), D19S916 (10.7%), D19S568 (1.8%) and D19S865 (3.6%). Loss of hetrozygosity in D19S413 predicts the relation between variation in this region and the disease.
Discussion: Our findings showed an average of 13.9% LOH in FNMTC cases. Among the five major microsatellites, D19S413 was the most informative for LOH analysis of FNMTC. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| BRIEF REPORT |
|
|
|
 |
Rhizomelic chondrodysplasia punctata: A missed opportunity for early diagnosis |
p. 344 |
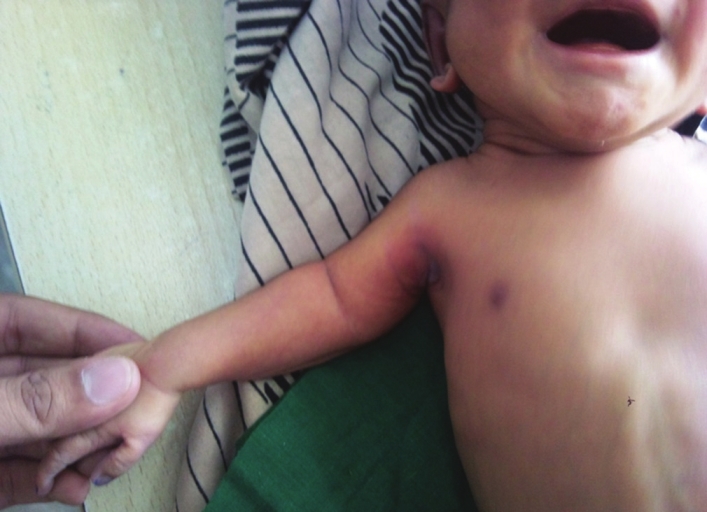
Nanda Chhavi, Sankar Prashanth, Chandrasekaran Venkatesh, Kadirvel Karthikeyan
DOI:10.4103/0971-6866.107990 PMID:23716944A male neonate was born with rhizomelic shortening of limbs. Skeletal radiograph showed punctate calcification of epiphysis of humerus, femur, and tibia. The diagnosis and a brief review of literature pertaining to the condition with emphasis on antenatal diagnosis and counseling are being reported. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| CASE REPORTS |
|
|
|
 |
Novel mutation in an Indian patient with Methylmalonic Acidemia, cblA type |
p. 346 |
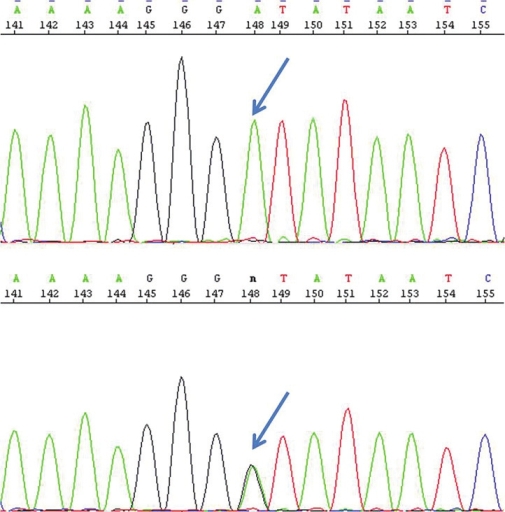
Katta Mohan Girisha, Aroor Shrikiran, Abdul Mueed Bidchol, Osamu Sakamoto, Puthiya Mundyat Gopinath, Kapaettu Satyamoorthy
DOI:10.4103/0971-6866.108025 PMID:23716945We report on a girl with methylmalonic acidemia, cblA type with a novel homozygous mutation and describe the clinical phenotype and response to therapy. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
An uncommon case of an adult with del(5)(q) in acute lymphoblastic leukemia |
p. 349 |
E Venkataswamy, Ashwini R Nargund, Shilpa Prabhudesai, Geeta V Patil, J Chandra Rao, Vidya H Veldore, Shekar Patil, Amit Verma, Rashmita Sahoo, BS Ajaikumar, Prasannakumari
DOI:10.4103/0971-6866.108028 PMID:23716946Del(5)(q) is a common chromosomal abnormality with favourable prognosis in Myelodysplastic Syndrome (MDS) and Acute myeloid leukemia (AML). However, del(5)(q) is also seen rarely in Acute lymphoblastic leukemia (ALL) and its significance remains poorly understood. We present here, a case report of diagnosis of an adult 75 year old patient of ALL with a cytogenetic abnormality of del(5)(q32). His clinical features, morphology and immunophenotyping findings were suggestive of T-ALL. Relevant literature has been reviewed and discussed. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
A new 48, XXYY/47, XYY syndrome associated with multiple skeletal abnormalities, congenital heart disease and mental retardation  |
p. 352 |
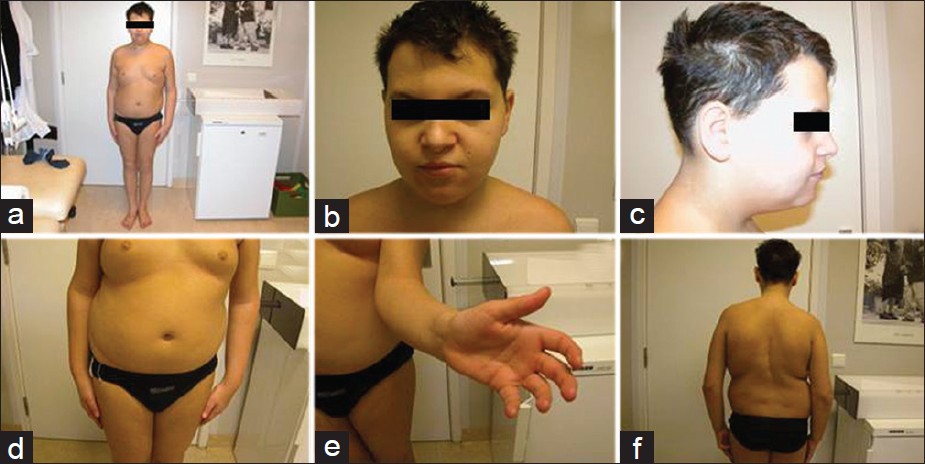
Leon Mutesa, Mauricette Jamar, Anne Cecile Hellin, Genevieve Pierquin, Vincent Bours
DOI:10.4103/0971-6866.108033 PMID:23716947While the XYY and XXYY syndromes have been several time described in patients, the combination of both syndromes in an individual is a rare event and may result in a severe phenotype. In the present observation, a boy with congenital scoliosis due to segmented thoracic hemivertebra associated with radioulnar synostosis and congenital heart disease is described. Chromosome G-banding and FISH analysis demonstrated a de novo mosaic karyotype 48, XXYY/47, XYY in this patient. To the best of our knowledge, this is the first report of a combination of XYY and XXYY syndromes. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
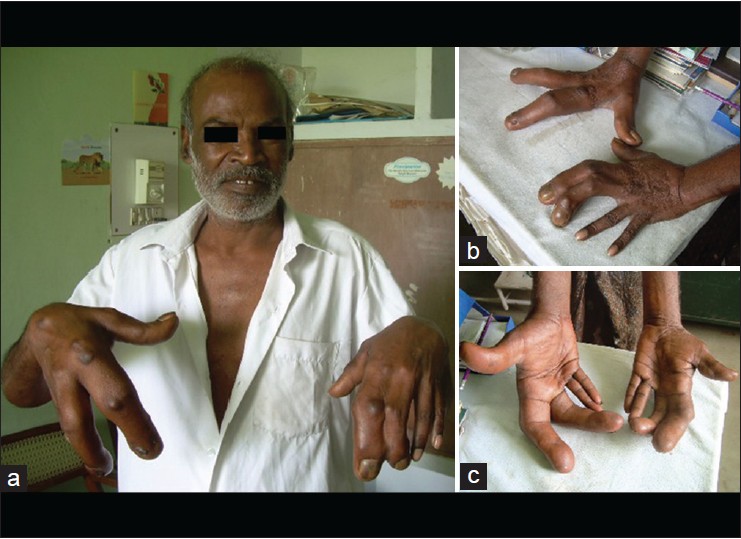 |
Proteus syndrome: A rare case report |
p. 356 |
Keerthi Talari, Praveen Kumar Arinaganhalli Subbanna, Deepak Amalnath, Subrahmanyam Dharanitragada Krishna Suri
DOI:10.4103/0971-6866.108036 PMID:23716948Proteus syndrome (PS) is a rare hamartomatous disorder characterized by various cutaneous and subcutaneous lesions, including vascular malformations, lipomas, hyperpigmentation, and several types of nevi. Partial gigantism with limb or digital overgrowth is pathognomonic of PS. We report a rare case of PS in a 50-year-old man who presented with inferior wall myocardial infarction and was incidentally detected to have hypertrophy of index and middle fingers of both the hands. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
A case of oligoasthenoteratozoospermia with AZFc deletion and persistent oxidative stress |
p. 359 |
Rajender Singh, Ahmad Mohammad Kaleem, Sankhwar Satya Narayana, Abbas Ali Mahdi
DOI:10.4103/0971-6866.108037 PMID:23716949Y-chromosomal microdeletions are associated with severe oligozoospermia or azoospermia. AZFc microdeletions have been always associated with severe oligozoospermia or azoospermia with a rare occurrence in individuals with other infertility phenotypes. We report here a rare case of an infertile man carrying AZFc deletion, whose semen picture is oligoasthenoteratozoospermia complexed with seminal oxidative stress. Anti-oxidant therapy could make no change in either oxidative stress biomarker levels of semen, seminal parameters or serum hormone levels. Therefore, oligoasthenoteratozoospermia in the present case correlates with AZFc deletion, and high content of abnormal sperm eventually might be responsible for persistently elevated reactive oxygen species levels. Understanding the function of genes in AZFc region could help decipher the exact cause of the phenotype in such cases. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
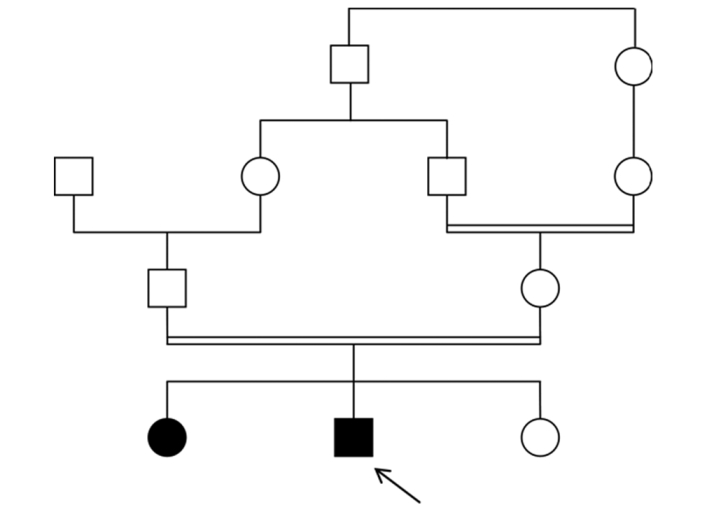 |
Renal amyloidosis due to familial Mediterranean fever misdiagnosed |
p. 363 |
Iman Hama, Ratbi Ilham, Naima Ouzeddoun, Zaitouna Alhamany, Radia Bayahia, Abdelaziz Sefiani
DOI:10.4103/0971-6866.108043 PMID:23716950Familial Mediterranean fever (FMF, MIM 249100) is an autosomal recessive disease affecting mainly patients of the Mediterranean basin. It is an autoinflammatory periodic disorder characterized by recurrent episodes of fever and abdominal pain, synovitis, and pleuritis. The major complication of FMF is the development of renal AA amyloidosis. Treatment with colchicine prevents the occurrence of recurrent seizures and renal amyloidosis. The disease is caused by mutations in the MEFV gene. We report here the cases of two unrelated patients, who have been late diagnosed with FMF complicated by renal amyloidosis. We focus on the importance of early diagnosis of FMF, both to start rapidly treatment with colchicine and avoid renal amyloidosis, and to provide genetic counseling to families. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
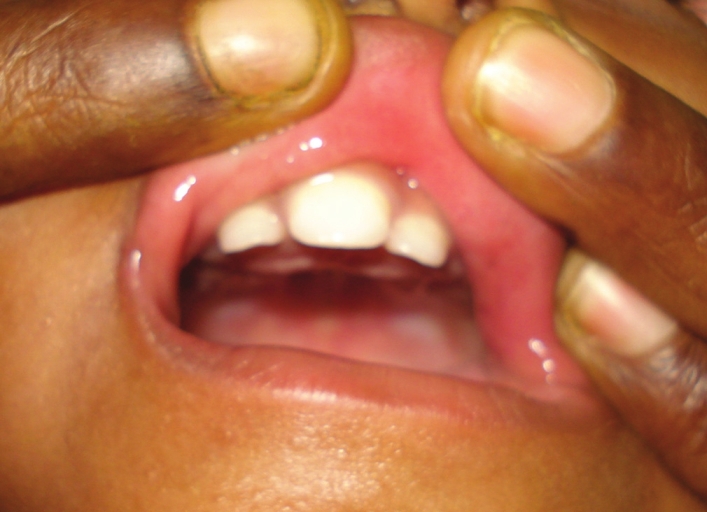 |
A new syndrome with overlapping features of Townes-brocks syndrome and single median maxillary central incisor syndrome |
p. 366 |
Thirunavukkarasu Arun Babu, Venkatesh Chandrasekaran, Sathish Balachandran
DOI:10.4103/0971-6866.108047 PMID:23716951A 14-month-old boy with overlapping features of Townes-Brocks syndrome (TBS) and single median maxillary incisor syndrome (SMMCIS) is being reported with brief review of the above syndromes and possible differential diagnosis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
A novel 11;18 translocation in a case of acute myeloid leukemia with maturation |
p. 369 |
G Sandhya Devi, Faiq Ahmed, Kavita Khadke, Sudha S Murthy, Senthil J Rajappa
DOI:10.4103/0971-6866.108050 PMID:23716952Acute myeloid leukemia with maturation (AML-M2) is associated with the 8;21 translocation. For the first time in an adult patient with AML-M2, a novel unbalanced translocation involving the short arm of chromosome 11 and long arm of chromosome18 with new breakpoints is presented. CD82 on band 11p11.2 and GATA 6 on 18q11.2 may play a role in the pathogenesis of de novo AML M2. The report with translocation (11;18)(p11.2;q11.2), as the sole cytogenetic abnormality provides more data on the leukemogenesis of de novo AML M2. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Seven nipples in a male: World's second case report |
p. 373 |
Tarang Goyal, SK Bakshi, Anupam Varshney
DOI:10.4103/0971-6866.108051 PMID:23716953We present a case of seven nipples in a 32-year-old male patient. The patient had two regular nipples along with five supernumerary nipples. Usually, supernumerary nipples develop along the two vertical "milk lines" which start in the arm pit on each side, run down through the typical nipples, and end at the groin. Our patient had six nipples which confirm to the "milk lines" and one nipple which was above the umbilicus in the midline and did not confirm to the "milk lines." To our knowledge, this is the second case report with seven nipples in the world. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
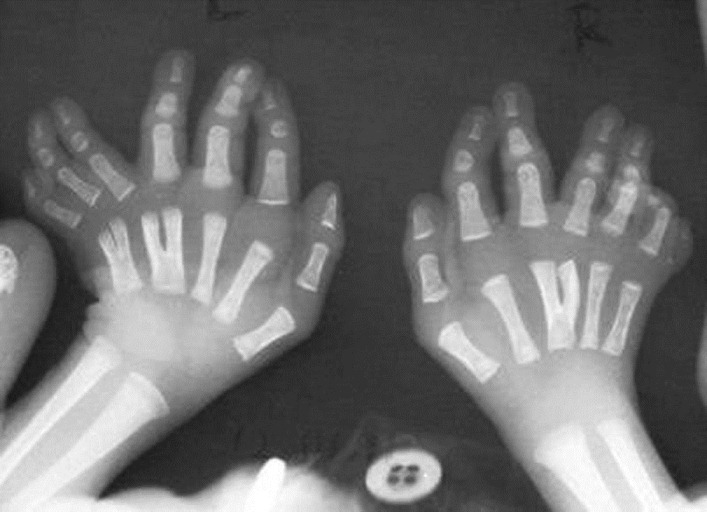 |
Orofaciodigital syndrome type-VI (Varadi-Papp syndrome) with several Y-shaped metacarpals |
p. 376 |
Pulak R Mahato, Shashi B Pandey
DOI:10.4103/0971-6866.108053 PMID:23716954Orofaciodigital syndrome type-VI (Varadi-Papp Syndrome) is a rare autosomal recessive disorder characterized by variable orofacial anomalies, central polydactyly of the hands, and cerebellar dysgenesis (mainly hypoplasia or aplasia of vermis, rarely Dandy-Waker anomaly). Here a case of Varadi-Papp syndrome with recurrent episodic tachypnea-apnea, minimal orofacial features, several Y-shaped metacarpals, and cerebellar vermis hypoplasia, diagnosed in the neonatal age, is reported for the first time in Indian literature. The importance of early accurate diagnosis of this rare disease for proper genetic counseling and prenatal case detection of pregnancy at risk is also emphasized as the prognosis is poor in almost all cases. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| LETTERS TO THE EDITOR |
|
|
|
|
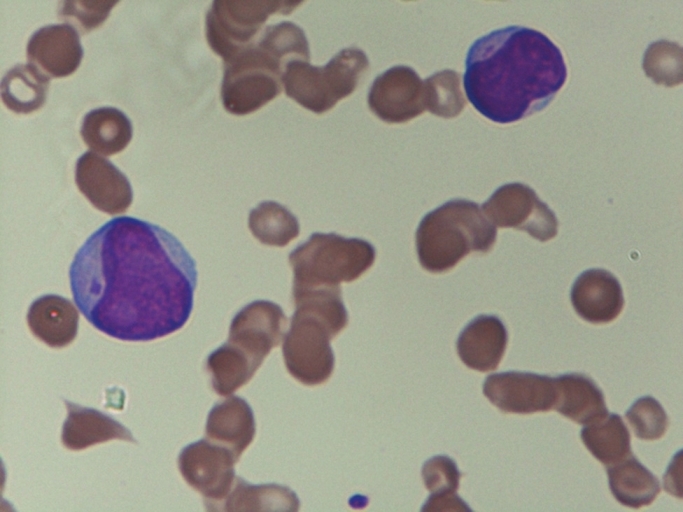
Octaploidy in idiopathic thrombocytopenia purpura: Is it incidental or causal? |
p. 379 |
Shantashri Vaidya, Babu Rao Vundinti
DOI:10.4103/0971-6866.108055 PMID:23716955 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
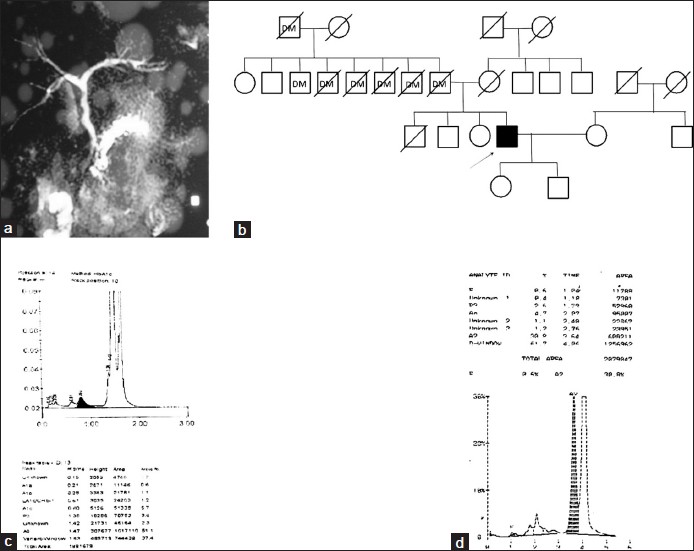 |
Chronic pancreatitis: A new pathophysiology |
p. 380 |
Shweta Singh, Ravindra Kumar, Gourdas Choudhuri, Sarita Agarwal
DOI:10.4103/0971-6866.108056 PMID:23716956 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|