|
 
 |
| REVIEW ARTICLE |
|
| Year : 2014 | Volume
: 9
| Issue : 2 | Page : 79-84 |
|
Bioequivalence comparison of generic drug DuLocap and brand drug Cymbalta
M Mudaliar
Department of Quality Assurnace and Regulatory Affairs, L. J. Institute of Pharmacy, Ahmedabad, Gujarat, India
| Date of Web Publication | 19-Aug-2014 |
Correspondence Address:
M Mudaliar
L. J. Institute of Pharmacy, Near Nagdev Kalyan Temple, Sanand Sarkhej Cross Roads, Ahmedabad 388 210, Gujarat
India
 Source of Support: None, Conflict of Interest: None  | Check |
DOI: 10.4103/9783-1230.139169

Abbreviated new drug application is required to submit to United States Food and Drug Administration for approval of the generic version of an innovator drug. This application is for (DuLocap) ® duloxetine hydrochloride delayed release (DR) capsules 60 mg manufactured by alpha pharmaceuticals. This application is an abridged application claiming essential similarity to the brand product that is, Cymbalta ® (duloxetine HCL DR capsules) 60 mg, mfg by Lily S.A, Spain and marketed by Eli Lilly, This is application is filed on the basis of para III (patent expires on June 11, 2013). Keywords: Abbreviated new drug application, Bioequivalence, Toxicology
How to cite this article:
Mudaliar M. Bioequivalence comparison of generic drug DuLocap and brand drug Cymbalta. J Med Investig Pract 2014;9:79-84 |
How to cite this URL:
Mudaliar M. Bioequivalence comparison of generic drug DuLocap and brand drug Cymbalta. J Med Investig Pract [serial online] 2014 [cited 2018 Aug 24];9:79-84. Available from: http://www.jomip.org/text.asp?2014/9/2/79/139169 |
| INTRODUCTION | |  |
Ceneric version of duloxetine hydrochloride is very much essential because depressive disorder is growing worldwide and the patients are continuously increases. Duloxetine is the best drug as an antidepressant and prescribe widely all over the world. The sale of duloxetine in 2012 was 1,158,353 US million dollar and ranked fourth among 100 top selling drugs of 2012. The price of Cymbalta (branded drug) is $125/30 capsule which is quite costlier to the patients of depressive disorder. The patents of Cymbalta will expire on June 11, 2103 holding by Lilly. After that, generic drug will come in the market. And the price of duloxetine HCL capsule will decreases and is affordable to every patient. Hence, the main objective is to provide a high quality duloxetine HCL capsule with cheap price. Delayed release (DR) dosage forms are designed to release the drug at a time rather than promptly after administration. The delay may be time based or based on the influence of physiological conditions like gastrointestinal tract pH. General information of Drug is shown in [Table 1] and [Table 2]. [1],[2],[3],[4],[5],[6]
Destroyed in the stomach or by intestinal enzymes
Known to cause gastric distress
Absorbed from a specific intestinal site or
Meant to exert local effect at a specific.
Duloxetine hydrochloride is a selective serotonin norepinephrine reuptake inhibitor (SNRI) currently indicated for the treatment of major depressive disorder (MDD), generalized anxiety disorder, diabetic peripheral neuropathic pain, and fibromyalgia. Duloxetine hydrochloride is an antidepressant drug. The degradation of this antidepressant drug in the acidic environment of stomach leads to sub therapeutic levels. In order to avoid this degradation and to bypass the acidic pH of the stomach, one of the proven approaches is a formulation of DR dosage forms.
| Selection of Drug: Duloxetine Hydrocloride Delayed Release Capsule | | |
Clinical overview
Duloxetine is classified as a SNRI. The claimed mechanism of action of duloxetine is based on the specific inhibition of both serotonin and norepinephrine (NE) reuptake, while it weakly inhibits dopamine reuptake and has no significant affinity for histaminergic, dopaminergic, cholinergic or adrenergic receptors. Dulocap 30 and 60 mg capsules contain enteric-coated pellets of the active substance duloxetine hydrochloride (+)-(S)-N-methyl-γ-(1-naphthalenyloxy)-2-thiophenepropanamine hydrochloride) equivalent to 30 and 60 mg of duloxetine. The recommended posology is 60 mg once daily with or without food. Dosages above 60 mg once daily, up to a maximum dose of 120 mg/day can be uptritated in evenly divided doses. Based on this posology, a combined package leaflet, including both strengths has been adopted. Duloxetine has been already approved for the treatment of stress urinary incontinence (SUI). The development program of duloxetine was initiated in the mid-1980s. The toxicology and toxicokinetic studies were performed in accordance with good laboratory practice regulations and except for the oldest studies, met Organization of Economic Co-operation and Development and Japanese Ministry of Health, Labor, and Welfare standards. Applicable ICH and CPMP guidance documents were also referred to during the development of the compound. All clinical studies with duloxetine have been conducted in accordance with good clinical practice and agreed ethical principles. General overview is shown in [Table 3].
Product development rationale
The rationale for the development and application for marketing approval of the duloxetine formulation is well-established. In vitro, duloxetine was an inhibitor of both serotonin and NE reuptake and a relatively weak inhibitor of dopamine reuptake. In addition, no significant affinity for histaminergic, dopaminergic, cholinergic or adrenergic receptors was evidenced. The effectiveness of duloxetine in the treatment of MDD is linked to its inhibition of presynaptic neuron reuptake of serotonin and NE in the central nervous system, resulting in elevated levels of serotonin and NE in the synaptic cleft, enhancing monoaminergic neurotransmission. Although no specific animal models for MDD are available, the in vivo pharmacodynamic studies provide indirect evidence for the potential clinical efficacy of duloxetine. At therapeutic range, duloxetine is not expected to pose a risk on central nervous system (CNS), smooth muscle, renal, immune, or gastrointestinal functions. Substance abuse is unlikely. The extent and scope of the documentation provided in this application are appropriate to support the nonclinical-pharmacology profile of duloxetine.
Overview of biopharmaceutics
Number of studies was conducted concerning absorption of duloxetine in several animal species. Duloxetine was well-absorbed in these species after gavage or dietary administration. After oral administration, duloxetine is well absorbed and extensively metabolized, Tmax being approximately 1.5 h in mice and rats. Absorption was found to be greatest from the duodenum and least from the stomach. In addition, pharmacokinetics of duloxetine were similar in fed and fasted male rats. Bioavailability (BA) was determined to be 21% in rats and 5% in dogs, probably due to extensive metabolism in the latter. No consistent gender differences in exposure occurred in dogs, although female rats tended to have higher exposure than male rats and male mice had greater values than female mice, but only at the highest doses tested. Even though, the area under the curve (AUC) values increased with increasing doses in mice, rats and dogs, these increases were not always proportional at the higher doses. Also, there was no evidence of accumulation with increasing duration of exposure.
Overview of clinical pharmacology
Primary pharmacodynamics (in vitro/in vivo)
A range of pharmacodynamic studies was performed with duloxetine concerning primary pharmacodynamics. Duloxetine is a combined SNRI (5-HT) and NE. Reuptake by nerve endings is thought to be a primary mechanism for removing monoamines from the synapse and terminating their effects at pre- and post-synaptic receptors. Therefore, blockade of reuptake may increase synaptic levels of monoamines and enhance monoaminergic neurotransmission.
In vitro studies
In vitro, duloxetine inhibits the uptake of 5-HT and NE. It weakly inhibits dopamine reuptake and has no significant affinity for histaminergic, dopaminergic, cholinergic or adrenergic receptors. Furthermore, duloxetine has relatively weak affinity in vitro for other neuronal receptors and binding sites. None of the various hydroxy-substituted compounds had appreciable affinity for the 5-HT, NE and dopamine transporters. The R (-) enantiomer of duloxetine, was also a potent inhibitor of 5-HT and NE reuptake.
Studies evaluating the pharmacodynamic potential of the possible metabolites and/or degradation products of duloxetine were performed. The 5- and 6-hydroxy analogs of duloxetine had relatively high affinity for human 5-HT and NE transporters in vitro with Ki values ranging from 1 to 18.4 nM. The 6-hydroxy-5-methoxy duloxetine metabolite had high affinity for human 5-HT transporters, whereas 5-hydroxy-6-methoxy duloxetine and the dihydrodiol isomers had low affinity for 5-HT and NE transporters. The 4-, 5-, and 6-hydroxy analogs of duloxetine, the dihydrodiol isomers, 5-hydroxy-6-methoxy duloxetine, 6-hydroxy-5-methoxy duloxetine and the conjugated compounds did not have appreciable affinity for neuronal receptors. Conjugates of the various hydroxy-substituted compounds had greatly reduced affinity for the 5-HT and NE transporters. None of the compounds had appreciable affinity for human dopamine transporters.
In vivo studies
The effectiveness of duloxetine in the treatment of MDD is linked to its inhibition of presynaptic neuron reuptake of serotonin and NE in the central nervous system, resulting in elevated levels of serotonin and NE in the synaptic cleft, enhancing monoaminergic neurotransmission. No specific animal models for MDD are available. Hence, the in vivo pharmacodynamic studies provide indirect evidence for the potential clinical efficacy of duloxetine. However, a forced swimming test, a behavioral test in rodents, is thought to predict the clinical activity of antidepressants. Duloxetine was investigated in several animal models measuring activities in noradrenergic and serotonergic neural systems and in "animal models" for depression. Duloxetine's activity in the behavioral assays indicates enhancement of 5-HT and NE neurotransmission, and activity in antidepressant models were in general agreement with the values obtained for blockade of ex vivo binding and increases in extracellular monoamines. Furthermore, it was investigated whether duloxetine produced an anticholinergic action. Duloxetine dose-dependently reduced tetrabenazine-induced ptosis in mice (ED50 of 4.0 mg/kg) and in rats (ED50 of 14.0 mg/kg). Reserpine-induced hypothermia was significantly reduced by duloxetine with an ED50 of 12.1 mg/kg. When duloxetine (12.5-100 mg/kg p.o.) and 5-hydroxytryptophan (80 and 100 mg/kg i.p.) were administered simultaneously to mice and rats, head movement behavior and tremor were observed. Duloxetine (ED50 of 27.6 mg/kg) reduced the immobility in the forced swimming test in mice. A dose-dependent (10-40 mg/kg) reduction in immobility by increasing the frequency of climbing behavior was observed in the forced swimming test in rats. A weak anticholinergic action was demonstrated at 25-50 mg/kg of duloxetine, but not at 100 and 200 mg/kg.
Secondary pharmacodynamics
Duloxetine demonstrated activity in nonclinical models of pain and effects on lower urinary tract function, as a consequence of its mechanism of action, via an enhancement of serotonin and NE neurotransmission. Duloxetine suppressed food consumption in mice and rats, and suppressed alcohol consumption in selectively inbred high-alcohol-drinking rats, in a dose dependent manner.
Overview of efficacy
Results of four initial Phase 2 studies (studies F1J-EW-E001, F1J-MC-HMAG, F1J-MC-HMAH, and F1J-MC-HMAI) were provided. Assessed the efficacy of low doses of duloxetine (ranging from 5 to 30 mg QD) in the treatment of MDD. Three out of them were double-blind, randomized and placebo-controlled studies and the other one was open-labeled and noncontrolled study. These studies failed to demonstrate a statistically significant difference of duloxetine over placebo for the primary endpoint, although showed some effect when considering some secondary endpoints. Hence, these initial dosages of duloxetine were considered as to be in the lower-bound of the effective range and additional "dose finding" studies were performed using higher doses (up to 160 mg/day). The two proof of concept studies HMAQa and HMAQb (powered 65%) also examined the doses. These were identical parallel group, double-blind, forced-titration active- and placebo-controlled studies comparing duloxetine titrated from 20 mg to 60 mg BID with placebo over 8 weeks of acute treatment. A fluoxetine 20 mg QD arm was used as an internal active comparator standard. The primary efficacy evaluation in these studies was the comparison between duloxetine 60 mg BID and placebo at endpoint using mean change analysis of the 17-item Hamilton Depression Rating Scale (HAMD17) total score. There were not statistically significant differences between fluoxetine and placebo for the mean change in HAMD17 total score in any of the studies. Duloxetine was numerically, but not statistically significantly, superior to placebo on the primary analysis. Only study HMAQa showed statistically significant differences for some of the secondary endpoints (HAMD17 Maier subscale, Clinical Global Impression (CGI)-severity, CGI-improvement, patient global impression-improvement, and SF-36 mental component summary and mental health subscales). The applicant concluded that only study HMAQa supports the efficacy of duloxetine 60 mg BID in the treatment of patients with Diagnostic and Statistical Manual of Mental Disorders-IV defined MDD. In the view of the Commitee of Human Medicinal Product (CHMP), the results of these studies are inconclusive, as neither duloxetine nor fluoxetine showed a significant greater effect than placebo. The applicant went on studying the efficacy of different dosages of duloxetine during the Phase 3 development program. In these studies duloxetine 20 mg BID, 40 mg BID, 60 mg BID and 60 mg QD were used. Based on the results of these studies, the applicant recommends the use of duloxetine 60 mg QD as the starting and the effective maintenance dose in the treatment of MDD. The main argument given by the applicant to support this dosage was that 60 mg QD was the only dosage that demonstrated superiority over placebo in the three studies (two acute and the relapse prevention study) in which it was tested (in contrast to that seen for the remaining dosages for which superiority over placebo was only seen in a half of the studies in which were tested) and that the effect size observed was better or comparable to other duloxetine doses tested and paroxetine 20 mg. However, duloxetine 60 mg QD (HMBH a and b) was not compared with any active control or to other duloxetine regimens.
Overview of safety
The potential of duloxetine to alter cardiovascular, CNS, smooth muscle, immune, and gastrointestinal motility functions, as well as its potential for substance abuse was examined as part of the safety pharmacology dossier provided.
Cardiovascular effects
In vitro, duloxetine had no effect on the function of the smooth and cardiac muscles at concentrations of 10-6 M and lower, while antagonistic effects were observed at concentrations of 10-5 and above. Duloxetine did not affect smooth or cardiac muscle function of rat and guinea pig at concentrations of 1 nM-1 μM. Cardiac ion channel results, together with QT measurements from dogs and humans would not indicate an arrhythmogenic risk with the use of duloxetine.
Central nervous system effects
Duloxetine would not be predicted to substantively affect CNS. It did not adversely affect CNS function of mice after single oral doses 3 mg/kg. At higher doses (10 and 30 mg/kg) an increase in hexobarbital-induced time sleep was observed, indicating that duloxetine may produce CNS depression or interfere with hexobarbital metabolism. Moreover, an increase in seizure threshold in electroshock-induced convulsions was observed at the highest dose. Comparable changes have been obtained following multiple-dosing (5 days). Mydriasis has been reported in association with duloxetine treatment.
Gastrointestinal, renal, and immunological effects
There were no important effects on gastrointestinal motility or immune functions in mice. Regarding renal function, no effects on urine volume were observed in rats based on the studies provided by the applicant. However, a slight elevation of serum potassium levels and an increase in sodium excretion were observed with 10 and 20 mg/kg doses.
Potential for abuse
The abuse potential of duloxetine was evaluated in monkeys and rats; duloxetine demonstrated the above mentioned CNS effects in rats, but not in rhesus monkeys up to 64 mg/kg doses. Duloxetine also did not demonstrate cross physical dependence potential with barbital or physical dependence-producing potential. As such, duloxetine would not be expected to pose a risk for substance abuse.
Based upon the results of these studies, therapeutically relevant doses of duloxetine would not be predicted to substantively affect CNS, smooth muscle, renal, immune, or gastrointestinal functions, neither would be expected to pose a risk for substance abuse. Potential secondary pharmacologic reactions at clinical doses would be limited to potential increases in pulmonary pressure, pulmonary vascular resistance, and respiratory rate. However, these effects are attributable to the known actions of NE and serotonin (Brunner and Gross 1979; Garattini and Valzelli 1965; Weiner 1985), and were only observed in anaesthetized animals.
Nonclinical summary
Only limited metabolism and excretion were evaluated in the monkey, since the dog had been chosen as the nonrodent specie for use in the toxicology program. Radiolabeled drug was administered in the pharmacokinetic, metabolism, excretion, and tissue distribution studies. The plasma protein binding of 14C-duloxetine has been determined in mouse, rat, dog, and human plasma. Additional studies have investigated the placental transfer of 14C-duloxetine in rats and the excretion of 14C-duloxetine into milk of lactating rats. [7]
Clinical summary
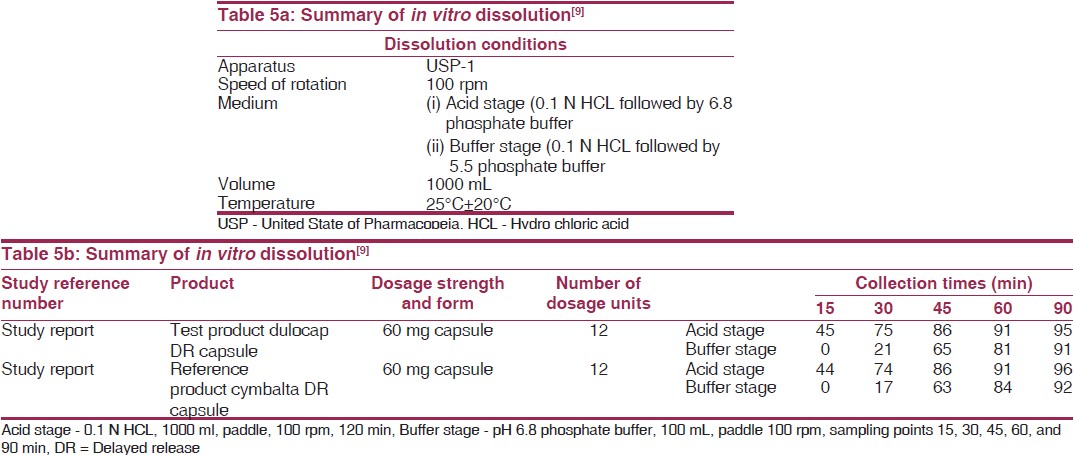
The pharmacokinetic and pharmacodynamic characteristics of duloxetine have been assessed in healthy subjects, special populations, and patients with either MDD or SUI. The pharmacokinetic data were available from a total of 294 subjects, including healthy volunteers, subjects with end-stage renal disease (ESRD), and subjects with cirrhosis in 21 clinical validation of drug is shown in [Table 4] and [Table 5], pharmacology studies and at least 421 patients in four Phase 2 and Phase 3 clinical trials.
Pharmacokinetics
Duloxetine is a chiral compound. The S-enantiomer seems to be the one that has been developed by the applicant. Since the in vitro pharmacologic activity of the two enantiomers is similar, interconversion, should it occur would be postulated to result in no or only slight changes in the in vivo therapeutic effect. Moreover, since the assay was not specific to a single enantiomer, the pharmacokinetics of duloxetine would reflect the combined data on both enantiomers. The pharmacokinetics of duloxetine have been characterized in healthy men and women over a single dose range of 20-60 mg and over a multiple-dose range of 40-160 mg/day. For the majority of the studies, duloxetine concentrations were determined using validated high performance liquid chromatography tandem mass spectrometry (HPLC/MS/MS) methods. The assay was validated over the concentrations range of 0.5-100 ng/mL with higher concentrations obtained after dilution of the samples. In some studies, the two major metabolites of duloxetine - the glucuronide conjugate of 4-hydroxy duloxetine and the sulfate conjugate of 5-hydroxy, 6-methoxy duloxetine - were determined. The metabolite concentrations were obtained using a validated HPLC/MS/MS method over the concentration range of 1-1000 ng/mL. The overall relative standard deviation (a measure of precision) and absolute relative error (a measure of accuracy) were >10% for both assays. Duloxetine and its two major metabolites were found to be stable in plasma stored frozen for the length of time that the study samples were stored under similar conditions.
Absorption
Results from mass balance studies indicate a fairly high extent of absorption (>70%) of duloxetine. Duloxetine is a DR formulation (capsules containing enteric-coated pellets), and as a result, Tmax occurs, at about 6 h. The absolute oral BA was estimated to a mean of 50%, but individual results ranged between 32 and 80%, respectively. Bioequivalence between the clinical trial and the marketing formulations of duloxetine was demonstrated using a 60 mg dose. This is acceptable, since all strengths of the final formulation contain the same enteric-coated pellets. When administered with a high-fat meal, there was a 6% decrease in Cmax and an 11% decrease in AUC 0-∞ . The absorption was delayed by food, with an increase in Tmax of about 4 h. A specific dose recommendation with respect to food intake is not considered warranted based on this. Analysis of result is shown in [Table 6].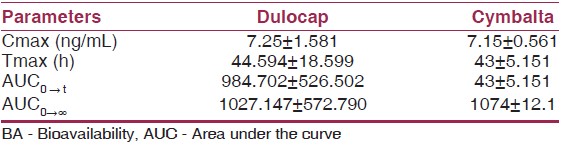 | Table 6: Comparison and analyses of results across studies and summary of BA studies[8]
Click here to view |
Distribution
Duloxetine has an extensive tissue distribution and is highly bound to plasma proteins. Different figures have been reported, but the degree of binding was generally above 95%. Duloxetine is bound both to albumin and to α1-acid glycoprotein (AAGP). Since AAGP is a protein with variable plasma concentrations, within and between patients, e.g. due to disease states like infections, this could be a source of variability in the pharmacokinetics of duloxetine. The urinary metabolites of duloxetine were moderately bound to plasma proteins and this binding was unaffected by the presence of duloxetine. Protein binding is not affected by ESRD or by hepatic impairment, and is independent of duloxetine concentration.
Elimination
Duloxetine is predominantly eliminated in the urine after undergoing extensive metabolism to numerous metabolites that are pharmacologically inactive. Comparison of the metabolite-to-parent ratio values after intravenous and oral duloxetine reveal that a larger fraction of the drug is metabolized upon oral administration, likely reflecting first-pass metabolism. The major biotransformation pathways involve oxidation in the naphthyl ring, followed by further oxidation, methylation, and conjugation. The two major circulating metabolites of duloxetine are the glucuronide conjugate of 4-hydroxy duloxetine and the sulfate conjugate of 5-hydroxy, 6-methoxyduloxetine. The cytochrome P450 isoenzymes involved in the oxidative metabolism of duloxetine seem to be CYP1A2 and CYP2D6 based on in vitro and in vivo data. Both CYP1A2 and CYP2D6 were identified as participants in the hydroxylation at 4 and 5 positions, the two dominant routes of duloxetine metabolism. Should the activity of one of CYP1A2 and CYP2D6 be absent or decreased in a subject, the other enzyme would be available to metabolize duloxetine. No unchanged duloxetine is excreted in the urine. In vitro studies indicate that neither of these metabolites contributes to the pharmacologic activity of duloxetine. Hepatic impairment results in a decrease in the formation of the metabolites, which leads to higher plasma concentrations of duloxetine. In contrast, renal impairment (ESRD treated with chronic hemodialysis) reduces their rate of elimination as evidenced by the longer t values of the metabolites.
Summary of biopharmaceutic studies and associated analytical methods
A selective, high sensitive and high throughput liquid chromatography-tandem mass spectrometry (LC-electrospray ionization-MS/MS) method has been developed and validated for the chromatographic separation and quantitation of duloxetine in human ethylenediaminetetraacetic acid plasma using fluoxetine (internal standard [IS]) as an IS. Analyte and IS were extracted from human plasma by liquid-liquid extraction using methyl-tert-butyl ether-n Hexane (80:20).The eluted samples were chromatographed on X-terra RP8 (50 4.6 mm, 5 μm particle size) column by using mixture of 30 mM ammonium formate (pH −5.0 ± 0.05) and acetonitrile as an isocratic mobile phase at a flow rate of 0.40 mL/min and analyzed by mass spectrometer in the multiple reaction monitoring using the respective m/z 298.08→154.0 for duloxetine and 310.02→148.07 for IS. The linearity of the response/concentration curve was established in human plasma over the concentration range 0.100-100.017 ng/mL. The lower detection limit (LOD, S/N > 3) was 0.04 ng/mL and the lower limit of quantization (LOQ, S/N > 10) was 0.100 ng/mL. This LC-MS/MS method was validated with intra-batch and inter-batch precision of 5.21-7.02. The intra-batch and inter-batch accuracy was 97.14-103.50 respectively. Recovery of duloxetine in human plasma is 80.31% and ISTD recovery is 81.09%. The main pharmacokinetic parameters were Tmax (h) = (7.25 ± 1.581), Cmax (ng/mL) (44.594 ± 18.599), AUC 0→t = (984.702 ± 526.502) and AUC 0→∞ , (1027.147 ± 572.790), respectively. [8]
| Conclusion | | |
Based on the dissolution profile of DuLocap is matched with innovator drug Cymbalta. Soon after expiry of Cymbalta, high quality generic drug is manufactured and is affordable.[9]
| References | | |
| 1. | Duloxetine. Available from: https://www.en.wikipedia.org/wiki/Duloxetine. [Last accessed on 2013 Feb 01]. 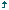
|
| 2. | Cymbalta ® : Official Site. Available from: http://www.cymbalta.com/Pages/index.aspx. [Last accessed on 2013 Feb 04].
|
| 3. | Rx list: The internet drug index. Available from: http://www.rxlist.com/cymbalta-drug.htm. [Last accessed on 2013 Feb 05].
|
| 4. | Cymbalta for fibromyalgia treatment. Available from: http://www.webmd.com/fibromyalgia/guide/cymbalta-for-fibromyalgia-treatment. [Last accessed on 2013 Feb 05].
|
| 5. | Drugs and Medications-Cymbalta Oral. Available from: http://www.webmd.com/drugs/drug-91491-Cymbalta+Oral.aspx?drugid=91491. [Last accessed on 2013 Mar 01].
|
| 6. | Medicinenet. Available from: http://www.medicinenet.com/duloxetine/article.htm. [Last accessed on 2013 Mar 01].
|
| 7. | European public assessment report. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR__Scientific_Discussion/human/000572/WC500036776.pdf. [Last accessed on 2012 Dec 16].
|
| 8. | Reddy. Development and validation of a LC/MS/MS method for the determination of duloxetine in human plasma and its application to pharmacokinetic study. E J Chem 2012;9:899-91.
|
| 9. | Pallavi. Formulation, development and evaluation of delayed release capsules of duloxetine hydrochloride made of different enteric polymers. Int J Drug Dev Res 2012;4:117-29.
|
[Table 1], [Table 2], [Table 3], [Table 4], [Table 5], [Table 6]
|









 Search Pubmed for
Search Pubmed for