Localization of Receptor-Mediated Signal Transduction Pathways: The Inside Story
Abstract
Receptor tyrosine kinases such as the epidermal growth factor receptor (EGFR) elicit proliferation, migration, and differentiation in a wide spectrum of cell types through various signal transduction pathways. These activities are attenuated by receptor internalization, intracellular trafficking through endosomes, and degradation in lysosomes, resulting in decreased receptor expression. However, there is now considerable evidence that EGFRs continue to signal in endosomes, forcing us to reevaluate the outcomes of receptor trafficking. An exciting revelation is that internalized receptors extend some signaling activities but not others, suggesting that certain responses, such as cell motility, must be mediated at the cell surface. Still, only when the effects of decreased receptor populations and signaling compartmentalization are integrated can we hope to understand and manipulate receptor function at the molecular level.

Introduction
Signal transduction through the epidermal growth factor (EGF) receptor (EGFR) or other receptor tyrosine kinases (RTKs) involves a series of dynamic, reversible processes that occur on different time scales. Receptor–ligand association is controlled by the off-rate of the ligand under low ligand concentrations, with the high affinity binding of most RTK ligands (Kd ∼ 1 nM) requiring minutes to achieve steady-state. Upon ligand binding, a series of rapid steps occur. RTKs form homo- or heterodimers with other receptors, activating their intrinsic kinase activity. Multiple tyrosine autophosphorylation sites in their cytoplasmic tails are modified in dynamic fashion [e.g., up to five sites on the EGFR are phosphorylated in vivo, and up to nine sites on the platelet derived growth factor (PDGF) β-receptor (PDGFR) are phosphorylated], with phosphates added and removed at various rates. These phosphotyrosine residues can then engage specific cytosolic signaling proteins that contain modular Src-homology 2 (SH2) and phosphotyrosine-binding (PTB) domains. To a large extent, the degree of phosphorylation at each autophosphorylation site and the accompanying affinities for various signaling proteins determine the receptor’s capacity for activating different signaling pathways in a given cell type. This “decoration” of a ligated receptor for intracellular signaling transpires within seconds or less, rapidly achieving a pseudo-steady-state with respect to the number of active receptors.
Three major pathways have dominated RTK signaling research [reviewed in (1, 2)]. The first to be identified involves the activation of phospholipase C (PLC)–γ, which hydrolyzes the lipid phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2]. This reaction produces 1,2-diacylglycerol in the plasma membrane, a potent activator of protein kinase C (PKC), and cytosolic inositol-(1,4,5)-trisphosphate [Ins(1,4,5)P3], which stimulates release of intracellular calcium. PLC-mediated lipid hydrolysis also liberates PI(4,5)P2-sequestered proteins, some of which modify actin during cell motility. The second major RTK pathway is mediated by the small, membrane-anchored guanosine triphosphatase (GTPase) Ras. In its active, GTP-bound state, Ras initiates a cell proliferation cascade involving the sequential activation of the protein kinases Raf, mitogen-activated protein kinase kinase (MEK), and extracellular signal-regulated kinase (Erk), in addition to other effectors. Oncogenic mutations in Ras are associated with about a third of all human tumors. Finally, RTKs stimulate type I isoforms of phosphoinositide 3’-kinase (PI3K), a stable heterodimer of regulatory and catalytic subunits, which phosphorylate PI(4,5)P2 in cells to produce PI(3,4,5)P3. This lipid second messenger and its breakdown product, PI(3,4)P2, engage specific enzymes and adaptor proteins that contain pleckstrin homology (PH) domains, mediating pathways for cell motility and survival.
Not all proteins recruited by active RTKs perform a positive signaling function. SH2-containing protein tyrosine phosphatase (SHP)-1 and SHP-2 are activated through interactions with autophosphorylated RTKs; these and other phosphatases can modify RTK signaling in both negative and positive ways [reviewed in (3)]. More generally, prolonged EGFR signaling in the continuous presence of EGF is attenuated over several hours by means of enhanced receptor internalization, ultimately leading to proteolytic destruction of receptor and ligand molecules. Induced EGFR internalization is achieved by trapping receptor–ligand complexes within clathrin-coated pits in the plasma membrane, through interactions with proteins such as Eps15 that engage active receptors [reviewed in (4)]. After the coated pits invaginate and endocytic vesicles form, the clathrin-coat dissociates, and both membrane- and fluid-phase molecules associated with these vesicles are delivered to early endosomes. It is in these acidified structures (pH ∼ 6), which number about 100–300 in a typical cell, where molecules are sorted for either degradation or recycling to the cell surface. Molecules that continue along the degradation route encounter increasingly reduced pH as they transit through late endosomes (pH ∼ 5) and, ultimately, lysosomes (pH ∼ 4). These perinuclear organelles are distinguished by unique protein markers, and they harbor proteases activated at low pH that hydrolyze internalized proteins [reviewed in (5)].
Thus, at steady-state, a pool of intact receptors resides in internal compartments. This has stimulated numerous questions. Do internalized ligands remain associated with their receptors? If receptors remain ligated, do they engage in signaling activities? If so, which ones? On a per receptor basis, are those pathways activated by internal receptors to an equal, greater, or lesser extent than by cell surface receptors? What are the mechanisms that differentiate signaling in various compartments? As in other reviews on this topic (6–11), the evidence here is presented with a focus on EGFR, the most intensively studied receptor in this field.
Receptor Downregulation: The Price of Compartmentalization
As we learn more about the roles of internalized receptors in initiating or extending signal transduction, it is easy to forget an important outcome of receptor-mediated endocytosis, elucidated in early studies: the downregulation of receptor number through degradation in lysosomes. Cells stimulated with a high concentration of EGF typically lose over 90% of their surface EGFRs at steady-state (12–14). Though relevant for cell proliferation in culture and in physiological settings, this state takes hours to achieve, longer than the time scale of most biochemical signaling measurements. In the first thirty minutes of stimulation, there is a significant distribution of EGF–EGFR complexes to intracellular compartments, but receptor downregulation is not yet significant (Figure 1⇓). However, one cannot discuss receptor signaling in endosomes without first considering the direct implications for prolonged receptor function.

Compartmentalization of active EGFRs. Time courses of surface-bound (A) and internal (B) EGF in NR6 fibroblasts expressing the human EGFR (J.M.H., unpublished). At time zero, cells were stimulated with a constant concentration of EGF (0.07, 0.2, 0.7, 2, 7, or 20 nM, from lowest to highest receptor binding), and surface-bound ligand was dissociated in pH 3 buffer at the indicated time points. C. Time course of EGFR autophosphorylation, 20 nM EGF stimulation, adapted from (27). Receptor activation changes very little in the first 30 minutes of maximal EGF stimulation, whereas after 24 hours these cells exhibit significant receptor downregulation (14).
Maintenance of receptor–ligand association in endosomes affects receptor processing and is a prerequisite for signaling. The likelihood of this depends on the free concentration of ligand in endosomes, which can be quite high. For example, if a cell contains 6,000 ligand molecules in a collective endosomal volume of 10−14 L, this corresponds to a maximum free concentration of 1 μM. However, the receptor–ligand affinity can be reduced significantly in the lower pH environments found in early and late endosomes. As a case in point, the EGFR binds EGF or tumor growth factor-α (TGFα) with similar affinities at pH 7.4 but very different affinities at reduced pH. EGF remains tightly bound to EGFR receptor in early endosomes, whereas TGFα dissociates nearly completely. This binding disparity significantly influences the sorting of EGF and TGFα. In normal cells, internalized EGF and TGFα are each degraded, but only EGF marks EGFRs for degradation as well, whereas TGFα spares receptors for recycling to the cell surface (15–17). The result is that EGF generally effects a more significant decrease of EGFRs. As with TGFα, the trafficking of low-density lipoprotein (LDL), asialoglycoprotein, and other ligands that uncouple from their receptors in endosomes has established that the dominant route of unbound receptors is partitioning into recycling endosomal tubules, while the ligand in the endosomal lumen tends to be degraded (18, 19). Together, these results suggest that EGFRs are retained in endosomal vesicles in an occupancy-dependent manner, as confirmed through steady-state endosomal sorting studies (20, 21). Indeed, proteins such as sorting nexin 1 (SNX1), hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs), and Cbl have since been identified as proteins that specifically target internalized EGFRs for lysosomal degradation [reviewed in (22)]. With the interconnected relationships among ligand occupancy, signaling, and receptor retention in endosomes, it is apparent that signaling in endosomes and signal attenuation through receptor downregulation are two sides of the same coin (Box 1).
Directing Traffic.
Downregulation of cell surface receptors in response to high extracellular ligand concentrations is achieved through receptor internalization and endosomal sorting for degradation versus recycling. Because both processes rely on receptor-ligand association, there is a complex and intimate relationship between receptor downregulation and the ratio of active receptors inside the cell versus on the surface. Mathematical models are useful tools for the analysis of multiple receptor binding and trafficking processes; the steady state model used here melds aspects of two more complex models (103, 104). As depicted in panel A, a critical aspect of the model is the retention of receptor–ligand (R–L) complexes in both coated pits at the cell surface and in endosomal vesicles, through interactions with accessory proteins of limited number (21, 105). Values of the rate constants describing constitutive internalization, coated pit internalization, transit of recycling tubules, and maturation of endosomes are indicated in units of inverse minutes. In a single cell, coated pits retain a maximum of 5,000 R–L complexes, and half this number is retained when roughly 20,000 R–L complexes are present at the surface. In endosomes, 86% of the freely diffusing receptors partition to recycling tubules, whereas 66% of the free ligand partitions to vesicles. A maximum of 5,000 R–L complexes can be retained in endosomal vesicles, and half this number is retained when 3,000 R-L complexes are present in endosomes. These parameters are consistent with EGFR trafficking experiments in NR6 fibroblast variants [Figure 1⇑ and (14, 106)].
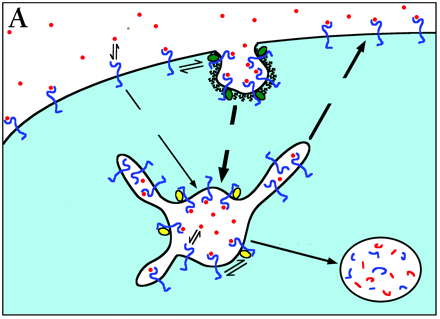
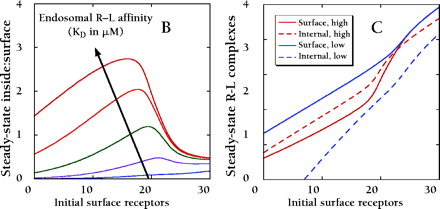
In a typical experiment, cells are stimulated with a saturating concentration of extracellular ligand. Panel B shows the predicted ratio of R–L complexes in endosomes relative to the cell surface at steady state, as a function of the initial receptor expression at the cell surface. Cells with low receptor numbers (≤ 104/cell) do not achieve high concentrations of ligand in endosomes, due to a low endocytic rate; the inside:surface R–L ratio is predicted to be suboptimal and sensitive to the ligand affinity at endosomal pH. In cells with very high receptor numbers (∼ 106/cell), a large number of R–L complexes both at the surface and in endosomes results in saturation of coated pit and endosomal retention processes, minimizing internalization and favoring recycling. This too results in a lower inside:surface ratio. Optimal compartmentalization of active receptors in endosomes relative to the surface is achieved at intermediate receptor numbers, which allow concentration of ligand in endosomes without significant saturation of trafficking processes.
The corresponding steady-state receptor numbers in the two compartments are shown in panel C for the highest and lowest endosomal receptor-ligand affinities depicted in panel B. Note that a ligand that dissociates in endosomes yields a higher number of R–L complexes at the surface, due to enhanced receptor recycling. At high receptor expression levels, this advantage disappears because the endosomal retention mechanism is saturated.
Compartmentalized Regulation of Receptor-Specific Properties
Given the sequence of events that follow receptor ligation, the maintenance of receptor–ligand association is only the beginning of the endosomal signaling story. Autophosphorylation of internalized receptors and their binding and potential phosphorylation of cytosolic signaling proteins can be altered relative to their cell surface counterparts (Figure 2⇓). This would allow the cell to regulate the magnitude and selectivity of signaling pathway activation through occupied receptors in various compartments.
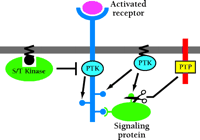
Compartmentalized regulation of receptor activities. Receptor-specific properties such as the pattern of receptor and signaling protein phosphorylation can be altered by endosome localization in a number of ways. Differential recruitment and/or activation of serine-threonine (S/T) kinases, which can phosphorylate the EGFR to inhibit its protein tyrosine kinase (PTK) activity, could change the extent and perhaps the specificity of substrate phosphorylation. Trafficking of other membrane-associated PTKs and receptor protein tyrosine phosphatases (PTPs) can yield higher or lower concentrations of these enzymes in endosomes versus the plasma membrane and provide additional regulation.
The maintenance of EGFR autophosphorylation in endosomes during EGF stimulation has been observed in A431 cells, a human carcinoma cell line that has been used in many studies because of their exceptionally high expression of EGFR (> 106/cell). In these cells, EGF remains tightly bound to EGFR in early endosomes, with no apparent change in the extent or pattern of receptor autophosphorylation (23–26). The extent of receptor phosphorylation is also unaffected by EGF–EGFR internalization in NR6 fibroblasts (∼ 4x105 EGFR/cell); in contrast, TGFα internalization leads to nearly complete dephosphorylation of the receptor (27). The autophosphorylation states of EGF–EGFR complexes at the cell surface and in endosomes of rat liver cells were also investigated, in a series of papers by Bergeron and colleagues (28–30). They found that the kinase activity and autophosphorylation of surface receptors are rapidly desensitized (t1/2 ∼ 10 s), whereas receptors in endosomes retain maximal activity in this cellular context. When analyzing the phosphorylation of EGF–EGFR complexes, it is important to note that only Tyr1173 of the EGFR is typically autophosphorylated to a significant extent, making it difficult to quantitatively detect changes in phosphorylation of the minor sites. The importance of this point is underscored by the experiments of Emlet et al., who observed time-dependent changes in Tyr992 phosphorylation within various compartments of EGF-stimulated A431 cells (31).
With the identification of numerous cytosolic signaling proteins that associate with active EGFRs and other RTKs, the relative abilities of surface and internalized EGFRs to bind and phosphorylate such signaling proteins have also been investigated. In rat liver, Shc is recruited to both the plasma membrane and endosomal fractions, but endosome-associated Shc is notably hyperphosphorylated. This prolonged Shc phosphorylation at endosomal membranes (and/or in the cytosol) may extend activation of the Ras/Erk pathway after selective desensitization of surface receptors (32). Differences in the extents of phosphorylation of PLC–γ1, Shc, and the p85 subunit of PI3K were also observed in HeLa cells that overexpress a dominant-negative mutant of dynamin, a GTPase required for clathrin-mediated endocytosis, as compared with wild-type dynamin expressing cells (33). In contrast, in NR6 fibroblasts that express human EGFR, a quantitative analysis showed that active receptors at the surface and in internal compartments contribute equally to tyrosine phosphorylation of both PLC-γ1 and Shc (27, 34). In this cell line as well as in mammary epithelial cell lines, surface and internal EGFRs also show equal association with Shc (34, 35). Still other proteins, such as Eps8 and Cbl, significantly associate only with internalized EGFRs, at least in some cell lines (35).
As we continue to probe the signaling properties of surface and internalized RTKs, many questions remain (Figure 2⇑). Can changes in receptor–protein interactions be explained by differential phosphorylation of minor receptor autophosphorylation sites? When tyrosine phosphorylation of receptor or signaling proteins are altered in endosomes, is this caused by differential regulation of the receptor’s kinase activity? A large body of literature has implicated serine-threonine kinases such as PKC in the feedback desensitization of EGFR activity [reviewed in (36)]. Thus, although it is plausible that reduced EGFR desensitization in endosomes (as observed in rat liver) reflects a signaling role for internalized receptors, reduced desensitization may also reflect what internalized receptors are not doing. Surface receptors may be desensitized as a result of localized signaling pathways that only they can activate. Another potential mechanism affecting phosphorylation and receptor–protein interactions may be achieved through regulated trafficking of other membrane components, such as nonreceptor tyrosine kinases (Src, ErbB-2), and certain protein tyrosine phosphatases (LAR, PTPs, DEP-1) (3). Indeed, it is noteworthy that Src and ErbB-2 are localized in or traffic through endosomes in certain cells (37, 38).
Availability and Trarricking of Proximal Membrane Substrates: The Next Frontier
It is probably no coincidence that each of the three major RTK signaling pathways targets membrane-associated molecules for modification, after receptor-mediated recruitment of the relevant enzymes. PLC and type I PI3Ks act upon the lipid PI(4,5)P2, and the Ras guanine nucleotide exchange factor (GEF) Sos acts upon Ras•GDP; phospholipase D (PLD)-dependent hydrolysis of phosphatidylcholine is yet another example. There are many reasons why this is probably important. First, a membrane-associated molecule is concentrated relative to a receptor-recruited enzyme, yielding up to 100–1,000 times higher reaction rate per molecule. Thus, membrane recruitment alone would substantially boost signaling (39, 40). Second, the planar geometry, close packing, and slower mobilities of molecules in cell membranes, versus those in the cytosol, promote spatial patterning. This is relevant for the formation of membrane microdomains (see below) as well as spatial gradient sensing during directed migration (41). Finally, and most relevant to this review, trafficking and enzymatic activities can alter the relative concentrations of membrane-associated molecules in various cell compartments.
PI(4,5)P2: Restricted Access
PI(4,5)P2 represents about 3% of all phosphatidylinositol lipids in unstimulated cells, which in turn comprise a small fraction of all cell membrane lipids. Still, this translates to as many as 108 molecules of PI(4,5)P2 per cell (or ∼ 100 μM based on cell volume). This lipid acts as a substrate for both PLC–γ and PI3K in signaling pathways stimulated by RTKs. Are these pathways affected by receptor internalization, and if so, does availability of PI(4,5)P2 play a role? The evidence, outlined below and summarized in Figure 3A⇓, strongly suggests that PI(4,5)P2 localization restricts specific signaling pathways and other cell activities to the plasma membrane.
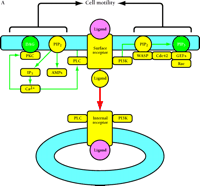
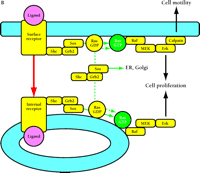
Compartmentalized activation of signaling pathways. A. Restricted localization of PI(4,5)P2. Surface and internalized receptors engage and phosphorylate PLC and PI3K, but the lipid substrate PI(4,5)P2 (PIP2) is not accessible to internalized receptors. Thus, only surface receptors mediate PIP2 hydrolysis by PLC, forming 1,2-diacylglycerol (DAG) and inositol (1,4,5)-trisphosphate (IP3) and releasing actin-modifying proteins (AMPs). IP3 releases intracellular calcium, which cooperates with DAG to activate protein kinase C (PKC). PI3K phosphorylation of PIP2 is also restricted to the cell surface. The product PI(3,4,5)P3 (PIP3) recruits many signaling proteins, including guanine nucleotide exchange factors (GEFs) that act upon the Rho family GTPases Rac and Cdc42. Rac•GTP mediates membrane ruffling, while Cdc42•GTP and PIP2 cooperatively activate Wiskott-Aldrich syndrome protein (WASP), which regulates actin polymerization. All of these signals play important roles in localized cell motility processes. PIP2 is also required for multiple stages of clathrin-coated pit endocytosis. B. (See next page.) Global activation of Ras and Erk. Ras•GTP loading is stimulated by receptor recruitment of Grb2–Sos complexes, mediated in many cell types by tyrosine-phosphorylated Shc. Both surface and internalized receptors bind to and phosphorylate Shc, recruit Grb2–Sos, and participate in Ras•GTP loading. It was recently confirmed that endosomes contain Ras, and Raf, MEK, and Erk are activated at each location. Activated Erk is released into the cytosol, and phosphorylation of cytosolic and nuclear Erk substrates is required for cell cycle progression. Erk activated at the plasma membrane is specific for activation of calpain, a calcium-dependent protease important for modulating cell adhesion and motility. Other recent evidence indicates that Ras can be activated in compartments that lack activated receptors, such as the ER and Golgi, presumably by Shc–Grb2–Sos complexes recruited from the cytosol.
In EGFR-expressing NR6 fibroblasts, quantitative measurements of EGF- and TGFα-stimulated PI(4,5)P2 hydrolysis showed that the PLC pathway is spatially restricted to the cell surface, with internal receptors contributing minimally, if at all, to this pathway (27). This was striking because, as noted above, surface and internalized EGF–EGFR complexes contribute equally to the tyrosine phosphorylation of PLC–γ1 in this cell line. In addition, green fluorescent protein (GFP) fusions of the PLC–γ1 tandem SH2 domains as well as full-length PLC–γ1 and PLC–γ2 are recruited to endosomes containing internalized EGF–EGFR complexes (42–44). Hence, the simplest explanation for the complete deficiency of internalized EGFRs in PI(4,5)P2 hydrolysis is that the lipid substrate is absent from endosomal membranes. In both rat basophilic leukemia and NIH 3T3 cells transfected with a GFP-labeled PH domain from PLC–γ1, which binds specifically to PI(4,5)P2 at the membrane [and Ins(1,4,5)P3 in the cytosol], association of the fluorescent probe with internal membranes was not noted (45, 46). Indeed, this fusion protein associated with the plasma membrane and some internal structures in NR6 fibroblasts expressing EGFRs and in A431 cells, but colocalization with internalized EGF was not detected (44). The same conclusion was reached using PI(4,5)P2-specific immunofluorescence in A431 cells (42). In the absence of growth factors, the GFP-PLC–γ1 PH domain fusion protein also failed to colocalize with internalized transferrin, indicating that the restriction of receptor-accessible PI(4,5)P2 to the plasma membrane is constitutive (44).
RTKs activate isoforms of PI3K that selectively phosphorylate PI(4,5)P2 in cells, producing PI(3,4,5)P3 [reviewed in (47, 48)]. Consistent with a lack of accessible PI(4,5)P2 in endosomes, a GFP fusion of the Akt PH domain, which binds specifically to PI(3,4,5)P3 [and PI(3,4)P2 derived from PI(3,4,5)P3], is recruited to the plasma membrane but not to endosomes upon EGF stimulation in the NR6 fibroblast system (44). Although EGFR-overexpressing fibroblasts do exhibit EGF-stimulated PI3K activity, it should be noted that EGF is generally a much poorer activator of PI3K than are PDGF, insulin, or other RTK ligands. In EGFR-overexpressing NIH 3T3 fibroblasts, a PI(3,4,5)P3 probe is recruited solely to the plasma membrane in response to EGF or PDGF stimulation (49); PI(3,4,5)P3-binding probes also fail to bind internal vesicles in 3T3-L1 adipocytes stimulated with PDGF or insulin (50). Thus, by segregating active receptors from accessible PI(4,5)P2, EGFR internalization impacts both PLC and PI3K pathways, and the constitutive lack of PI(4,5)P2 in endosomes suggests that this is the case for many signaling receptors.
The restriction of PI(4,5)P2 to the cell surface is not surprising when one considers the requisite roles of PLC and PI3K pathways in RTK-mediated cell motility (51, 52) (Figure 3A⇑). These pathways stimulate migration by affecting the dynamics of actin filaments in different ways, activities that must be limited to the cell cortex. During chemotaxis, lipid products of PI3K action are robustly polarized in the direction of the chemoattractant gradient, highlighting the importance of tightly localized signaling [reviewed in (53, 54)]. PI(4,5)P2 can also affect cell motility in more direct ways. It regulates plasma membrane tension through cytoskeletal attachment (55), and it engages in a costimulatory interaction with N-WASP, a regulator of actin polymerization (56, 57). With regard to the latter mechanism, and in light of the fact that PI(4,5)P2 levels change very little as a result of receptor stimulation, it has been effectively argued that PI(4,5)P2 simply acts as a signpost marking the plasma membrane (58). Thus, N-WASP may be cooperatively stimulated by two signals, by analogy to a “coincidence detector or a logical ‘AND’ gate” (59), in which the Rho family GTPase Cdc42 provides the magnitude and PI(4,5)P2 reinforces the location.
Interestingly, PI(4,5)P2 is intimately involved in receptor endocytosis as well, mediating clathrin-coated pit assembly and formation of endocytic vesicles through interactions with AP180, epsin, AP-2, and dynamin [reviewed in (60, 61)]. After all, these proteins must be properly targeted to the plasma membrane and not other locations. However, the role(s) of PI(4,5)P2 during the early stages of endocytosis also suggest how the lipid must be excluded from endosomes. Shortly after internalization of endocytic vesicles, the clathrin coat is disassembled, and it has been postulated that rapid PI(4,5)P2 clearance is required for this process. This hypothesis is supported by the apparent participation of synaptojanin isoforms, PI(5)-phosphatases enriched at clathrin-coated pits, in vesicle uncoating. Thus, PI(4,5)P2 may be consumed prior to fusion of endocytic vesicles with early endosomes. PIP(5)-kinase activity, which is regulated by signaling from PLD and Rho-family GTPases to maintain PI(4,5)P2 at high levels, would also need to be tightly regulated.
The lack of PI(4,5)P2 hydrolysis contributed by PLC–γ1 phosphorylated by internalized EGFRs indicates that the enzyme must be properly localized for activity. Receptor association, which could allosterically activate or provide continuous tyrosine phosphorylation to PLC–γ1 in the face of high phosphatase activity, along with high concentrations of PLC–γ1 adjacent to the membrane, are probably critical. This is certainly also the case for PI3K (47). It can be argued that PLC–γ1 phosphorylated by internal receptors might be recruited to the surface, in which case the maintenance of EGFR activity in endosomes would prevent a decrease of PLC activity. However, a mathematical analysis of the available data suggests that PLC–γ1 is effectively dephosphorylated when not bound to activated receptors (Box 2). Therefore, the phosphorylation of the receptor and its ability to bind proteins in endosomes may be irrelevant with respect to certain pathways, if specific membrane proximal substrates such as PI(4,5)P2 are not present.
A Favorable Exchange Rate?
RTKs bind to and phosphorylate cytosolic signaling proteins. For many of these, tyrosine phosphorylation affects protein function; Shc phosphorylation enables its interaction with Grb2, and phosphorylation positively influences the enzymatic activities of PLC–γ and PI3K. Once phosphorylated, the proteins may dissociate into the cytosol and diffuse about before binding again to an activated receptor. While in the cytosol, a phosphorylated signaling protein may be acted upon by cytosolic phosphatases. It is likely that phosphorylated proteins in the cytosol, unphosphorylated proteins recruited to the membrane, and phosphorylated proteins at the membrane can all be activated for signaling to different extents.
Here the term ”phosphorylated fraction” is taken to mean the fraction of a specific signaling protein in a given compartment that is phosphorylated. How does the phosphorylated fraction of a protein in the cytosol depend on the phosphorylated fractions associated with the plasma membrane and endosomes? Under what conditions can the phosphorylated fraction at endosomal membranes affect that of the plasma membrane? These questions have been addressed using mathematical modeling (107). The figure below depicts three compartments, plasma membrane (PM), cytosol (Cyt), and endosomes (EN) under various conditions; the height of the bars signifies the number of molecules in the compartment, and the color indicates the phosphorylated fraction (darker = higher fraction). In A, the PM and EN compartments recruit equal numbers of the signaling protein, whereas in B, the number in EN is increased. Under condition 1, the phosphorylated fractions in PM and EN are the same, and the signaling protein is not dephosphorylated significantly while in the cytosol. The phosphorylated fraction in Cyt thus takes on the fraction seen at the membranes and is unaffected by an increase in EN recruitment in B. In 2, the EN compartment produces a higher phosphorylated fraction. Again phosphatase activity is not significant in the cytosol, and the Cyt phosphorylated fraction is a weighted average of the fractions in PM and EN (compare A and B). The PM phosphorylated fraction is not affected by the EN because, in this case, proteins exchange with the cytosol very slowly compared to the rates of phosphorylation and dephosphorylation. However, under condition 3, in which PM and EN have the same respective kinase activities as in 2 but proteins exchange rapidly with the cytosol, the weighted average phosphorylated fraction is observed in all three compartments. In 4, 5, and 6, the conditions are as in 1, 2, and 3 respectively, except that cytosolic phosphatase activity is now high. Coupled with rapid exchange as in 6, phosphorylation can not be maintained in any of the compartments.
These conditions can be distinguished experimentally if one measures total tyrosine phosphorylation for various levels of total receptor activation and internal:surface receptor ratio. When such an analysis was performed for EGFR phosphorylation of PLC–γ1 and Shc in NR6 fibroblasts, the results were definitive. PLC–γ1 phosphorylation maps to condition 4, whereas Shc phosphorylation is uniquely described by condition 1.
Ras: Global Access
The three isoforms of the p21 Ras GTPase (H-, N-, and K-) are anchored in the membrane by lipid modifications. All are farnesylated at the C terminus, followed by further post-translational processing. H- and N-Ras are then palmitoylated at the endoplasmic reticulum (ER) and exported through the Golgi to the plasma membrane, whereas K-Ras does not receive further lipid modification and bypasses the Golgi (62, 63). Typically, greater than ninety-five percent of Ras is associated with cell membranes, and biochemical studies as well as fluorescence recovery after photobleaching experiments have shown that membrane association of Ras is stable (64). The majority of Ras is maintained in the inactive, GDP-bound state in unstimulated cells, courtesy of Ras GTPase-activating proteins (Ras GAPs). In response to RTK stimulation, the dissociation of GDP is stimulated by Ras GEFs such as Sos. This favors loading of the more abundant GTP from the cytosol onto Ras, increasing the percentage of Ras in the active state. Sos is constitutively associated with Grb2, an SH3-SH2-SH3-containing adaptor protein that can connect Sos to autophosphorylated receptors. EGFR phosphorylation of Shc provides an alternative process whereby Grb2–Sos are recruited to active receptors. Although the increase in Ras•GTP levels stimulated by EGF correlates quantitatively with an increase in the rate of GDP–GTP exchange in fibroblasts (34, 65), the possible regulation of GAP activity should not be overlooked. Indeed, the SH2 domains of p120 RasGAP bind directly to activated RTKs, and the localization of Gap1m is regulated by the binding of its PH domain to PI(3,4,5)P3.
The relative abilities of surface and internalized EGFRs to generate Ras•GTP have been assessed in the NR6 fibroblast system. As discussed above, receptor binding and phosphorylation of Shc are not altered in those cells; however, it is possible that membrane trafficking processes could yield higher or lower concentrations of Ras in endosomal membranes compared to the plasma membrane. Nonetheless, surface and internal receptors were equipotent in generating Ras•GTP (34). This study was the first to demonstrate activation of Ras by internalized receptors. Given the previous finding that only surface EGFRs engage in PLC-mediated PI(4,5)P2 hydrolysis in the same cells, it is apparent that cells can be selective with respect to the extension of specific signaling activities by internalized receptors.
Do these results shed light on how Ras GEF activity is regulated? There continue to be two schools of thought (Figure 3B⇑). One view holds that tyrosine phosphorylation of Shc and the subsequent formation of Shc-Grb2-Sos complexes is sufficient for increased Ras GEF activity. This mechanism is at least possible given the evidence that Shc phosphorylation persists in the cytosol between encounters with active receptors (32) (see also Box 2). The second view contends that Ras GEF activity must be recruited to the cell membrane where Ras is located, given the kinetic considerations discussed previously. It should be noted that the two mechanisms are by no means mutually exclusive, because the localization effect would synergize with any allosteric effects imparted by Shc association. However, if membrane localization is required for increased Ras GEF activity, then the evidence suggests that endosomal membranes and the plasma membrane contain equivalent concentrations of Ras, at least in NR6 fibroblasts.
Do endosomes contain Ras? Using expressed proteins tagged with GFP variants, Jiang and Sorkin recently observed colocalization of Grb2, Shc, and EGFR at the surface and in endosomes of three different receptor-overexpressing cell lines (66). Fluorescence resonance energy transfer (FRET) measurements confirmed that these molecules form complexes. Importantly, the endosomes also contained H- and K-Ras, both before and during EGF treatment. Although the guanine nucleotide-binding state of Ras may alter aspects of its trafficking, it is unlikely that the EGFR can enhance Ras internalization by dragging it along the endocytic route. It has long been known that Ras GEFs do not form stable complexes with Ras in the presence of GDP and GTP (67); rather, GEFs must promote exchange through a “kiss-and-run” mechanism. Indeed, FRET between labeled EGFR and Ras isoforms could not be detected (66). However, the study revealed that the ratio of endosome-to-plasma membrane localization was higher for H-Ras than K-Ras, indicating that at least one of these Ras isoforms does not experience bulk membrane trafficking. It was speculated that H-Ras might be delivered directly to endosomes from the Golgi, although differential localization and migration from plasma membrane microdomains may also explain the relative distributions of H- and K-Ras (68).
Another recent report suggests that Ras can also be activated in cell compartments that lack activated receptors. Chiu et al., who also employed live-cell imaging methods, found that Ras in the ER and Golgi, presumably in the process of palmitoylation and exocytosis, is activated in response to EGF stimulation in Cos-1 cells (69). This study strongly suggests that EGF–EGFR complexes do not need to be present in the compartment where Ras is activated. However, this does not necessarily mean that p(Tyr)-Shc–Grb2–Sos complexes do not need to be membrane localized. Lotti et al. reported that Shc is localized on ER membranes in unstimulated cells, and that EGF stimulation of cells redistributes Shc to the plasma membrane and endosomes (70).
It should be noted that the two recent studies using live-cell imaging to assess the localization and activation of Ras are not without caveats. First, many of the conclusions were based on experiments in which Ras was overexpressed and fused with GFP variants, which may alter Ras processing and function. In many cell types, Ras is endogenously expressed—between 10,000–50,000 molecules per cell—with only a small fraction in the GTP-bound state. Second, the presence of Ras•GTP was visualized using the modular Ras-binding domain (RBD) of c-Raf-1. In order to visualize this probe in the cytosol, the cell must express it at micromolar concentrations, but its affinity for Ras•GTP is in the low nanomolar range (71). Therefore, Raf-RBD will bind all available Ras•GTP until most of the probe is depleted from the cytosol. Because Ras GAPs must compete with the Raf-RBD to bind the Ras effector domain, the probe will alter the activation kinetics of Ras when expressed at detectable levels. Indeed, it was noted that the magnitude of FRET between labeled Ras and Raf-RBD did not change significantly in response to EGF stimulation, such that only the localization of the interaction could be assessed (66). Nevertheless, the evidence for Ras activation at locations other than the plasma membrane is compelling, and the imaging studies are in complete accord with the biochemical analyses with respect to the ability of internalized EGFR to activate Ras.
Plasma Membrane Microdomains: Regulated Access?
Over the past decade, our understanding of the structure and function of the plasma membrane has changed dramatically, in parallel with the profound advances in signal transduction research. We now appreciate that the membrane is not simply a continuous fluid bilayer in which lipids and proteins diffuse freely and randomly. Rather, the close packing of these molecules forces interactions that cause segregation of distinct microdomains. Clathrin-coated pits are but one example. Receiving even more attention recently are caveolae and lipid rafts, low-buoyant-density structures held together by cholesterol. Lipid microdomains remain enigmatic for two major reasons. First, although microdomains can be separated from the bulk, “disordered” plasma membrane, different low-density structures are difficult to separate from one another (72). Second, they are small enough (∼ 50 nm) to limit their characterization by fluorescence microscopy and other imaging methods. Therefore, the roles of microdomains in regulating receptor trafficking and signaling are currently controversial, as discussed in other reviews (7, 9).
Many RTKs (EGFR and PDGFR) and signaling molecules [including PI(4,5)P2, H-Ras, and Src] are apparently enriched in low-density microdomains, and this high concentration of key players has led to the hypothesis that caveolae and rafts foster signal transduction (73, 74). Moreover, since these structures are only found in the plasma membrane, microdomains could contribute to the compartmentalization of signaling. Ligand binding regulates the concentration of EGFR in microdomains, with bound receptors migrating out of microdomains. This release is apparently mediated by Src, which can phosphorylate caveolin-1 (75, 76), and Src activity also enhances the rate of EGFR internalization. In addition to regulating migration from caveolae, which may be a necessary precedent for clathrin-mediated internalization, Src may enhance internalization by promoting clathrin-coated pit assembly through phosphorylation of clathrin (77, 78). Recent studies by Hancock and colleagues have shown that H-Ras migrates from microdomains as well (68, 79, 80). Intriguingly, H-Ras migration is dependent on GTP loading of H-Ras, and mutations that prevent its migration inhibit interactions with Raf, PI3K, and Ras GEFs. K-Ras was not detected in lipid rafts or caveolae, and it is tempting to speculate that this affects the relative abilities of H- and K-Ras to enter endosomes.
Completing the Pathway: Activation of Downstream Kinases
As discussed above, the evidence strongly suggests that PLC and PI3K activities are restricted to the cell surface and depend on the availability of PI(4,5)P2. Accordingly, there is no evidence that enzymes such as Akt that engage lipid products of type I PI3Ks are activated at endosomal membranes (localization of the lipid product of PLC, 1,2-diacylglycerol, is a more complex issue because this species is also produced by PLD hydrolysis of phosphatidylcholine). Ras, on the other hand, can be activated in endosomes as well as at the plasma membrane, and there are a number of corresponding reports linking internalized EGFR to the activation of Raf, MEK, and Erk. However, this relationship is complicated by Ras-independent mechanisms that contribute to the activation of this cascade (81).
Erk activation was strongly inhibited in HeLa cells expressing dominant-negative dynamin, which blocks EGFR endocytosis, as compared to cells overexpressing wild-type dynamin (33). This implicates EGFR internalization as an important step in Erk activation, and analyses of G protein-coupled receptor and insulin receptor internalization and Erk activation using a variety of approaches to block endocytosis have led to the same conclusion (82–85). As additional evidence that Erk can be activated by EGFRs in endosomes, isolated endosomes from EGF-stimulated A431 cells activate Erk in cytosol extracts (86). However, the conclusion that EGFR internalization is required for full activation of Erk is not well supported. An EGFR variant that is defective in endocytosis activates Erk to the same extent as wild-type receptors (51). In one study, G protein-coupled-receptor ligands that do not stimulate receptor internalization activated Erk as well as their internalizing cousins. In that cell context, dominant-negative dynamin inhibited Erk activation in all cases, suggesting that receptor internalization is not the only outcome affected by dynamin inhibition (87). Indeed, using the HeLa cell line expressing dominant-negative dynamin, different results were found when comparing cells in which expression was induced versus uninduced, rather than comparing dominant-negative cells to cells overexpressing wild-type dynamin (88). Specifically, expression of dominant-negative dynamin had no significant effect on EGF-stimulated Erk activation. Above all, the fact that TGFα is only associated with EGFR at the cell surface, yet efficiently activates Erk to stimulate cell proliferation, is rarely mentioned and should be stressed. Based on all of the evidence, it is likely that both surface and internalized receptors are capable of activating the full Ras–Erk pathway, although the relative extents may differ across various cell–receptor systems. The apparently global activation and regulation of Ras–Erk may reflect its role in signaling cell cycle progression (Figure 3B⇑), with Erk translocation to the nucleus the critical step. In other words, there is no reason why such a pathway needs to be confined to the surface. It has even been speculated that endosome-associated Erk is protected from phosphatase activity in the cytosol, as a requirement for transit to the nucleus (89).
Activated Erk is also required for aspects of EGF-stimulated cell motility (90, 91), and thus, some Erk may need to be confined to the cell surface as in the case of PLC- and PI3K-mediated pathways. Can a cell distinguish between Erk activated at or near the plasma membrane versus other locations to affect the specificity of different cell responses? A recent paper suggests exactly this. EGF-stimulated, Erk-dependent activation of calpain protease activity is required for fibroblast migration. Exploiting the differential binding of EGF and TGFα to EGFR, Glading et al. found that only surface EGFR contributes to this pathway. They further showed that artificial targeting of Erk to the plasma membrane efficiently activates calpain, whereas cytosolic sequestration of Erk prevents calpain activation but not phosphorylation of a cytosolic Erk substrate (92).
Receptor Trafficking and Signaling an: Integrated View
One of the challenges in designing interventions to affect signaling by RTKs is that these pleiotropic receptors stimulate multiple cell responses. Thus, simply blocking a receptor–ligand interaction can have many functional outcomes. An alternative and perhaps more specific approach is to target intracellular signaling proteins for intervention. This is not a perfect solution, however, because a signaling pathway can impact responses mediated by many receptors in different cells, as well as multiple responses in the same cell. Recall that Erk is required for both proliferation and motility, and PI3K affects both directed migration and cell survival. Adding to the complexity is signaling crosstalk—the ability of multiple pathways to interconnect at signaling nodes. For example, PI3K is an effector of Ras•GTP, such that interventions modulating Ras activity will also impinge on PI3K. The compartmentalization of these signaling pathways and associated cell responses may offer another basis for intervention, by influencing the activity of receptors in endosomes relative to the cell surface (Figure 4⇓). This could be achieved by changing properties of the ligand [reviewed in (93)] or by targeting accessory proteins that influence receptor trafficking; however, receptor downregulation and ligand depletion, inherently linked to compartmentalization of signaling, are most often the dominant long-term effects (14, 94).
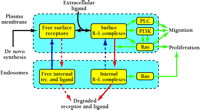
Integrated model of RTK regulation and signaling. Signal transduction hinges on the number of active receptors and their compartmentalization over time. Both are subject to receptor trafficking processes. Dashed arrows signify processes that occur at slower rates per receptor in normal cells; however, these processes become increasingly significant as the number of receptors increases, with saturation of the internalization and endosomal retention mechanisms. In addition to regulating the total number of receptors, trafficking processes may bias the specificity of receptor signaling. Surface receptors activate the PLC, PI3K, and Ras–Erk pathways, stimulating both cell migration and proliferation. The Ras–Erk and PI3K pathways engage in crosstalk interactions in both directions, and some of these may only manifest themselves at the plasma membrane, reinforcing cell motility signaling. Internalized receptors can activate the Ras–Erk pathway to extend the duration of cell proliferation signals. Interestingly, activated Ras can enhance receptor internalization through the GTPase Rab5 [reviewed in (22)], providing a negative feedback loop mediating receptor downregulation and a regulatory mechanism governing the balance of cell migration and proliferation signaling.
Know Thy Receptor Number
Much of the work on EGFR trafficking and signaling has utilized cells that express high levels of the receptor (> 105 molecules/cell). Although it may be argued that the results do not reflect normal cell physiology, many tumor cells do exhibit such high numbers of receptors (95, 96). How do high receptor levels contribute to cell transformation? At high concentrations of EGF, receptor downregulation is significantly curtailed owing to the saturation of both internalization and endosomal retention processes (Box 1). In this case, sorting of EGFR in response to EGF and TGFα become more alike, yet extracellular EGF is not depleted as rapidly as TGFα (14). This may explain why TGFα is such a potent autocrine factor in transformed cells; a continuous source of ligand is coupled with a receptor that cycles through endosomes and back to the surface (97, 98). Expression of large amounts of EGFR (greater than 105/cell) makes it likely that secreted TGFα will be captured by the secreting cell rather than by neighboring cells (99). High receptor expression also alters cell physiology at low ligand concentrations. Intracellular signaling proteins may be limiting, such that maximal signaling is achieved at very low receptor occupancy. In EGFR-overexpressing NR6 fibroblasts, both Ras•GTP generation and the rate of cell proliferation are saturated at submaximal receptor activation (14, 34). The difference is that receptor downregulation is not an issue at low receptor occupancy, because the receptor degradation rate is not significantly increased. Further, the cell can internalize enough empty receptors to maintain tight binding of internalized EGF. Finally, another consideration is the expression level of ErbB-2/Neu, an orphan receptor and cousin of EGFR that has been linked to numerous human cancers (100). By forming heterodimers with EGFR–ligand complexes, ErbB-2 serves as a potent signaling platform and, more insidiously, significantly interferes with EGFR downregulation (101).
In tumors where EGFR and/or ErbB-2 are overexpressed, receptor-antagonizing monoclonal antibodies and ErbB receptor family-specific kinase inhibitors have been proposed as therapeutic drugs [reviewed in (102)]. These will counter the effects described above; however, if intracellular signaling components are limiting, such interventions may have more dramatic effects on “normal” cells with more closely matched receptor-binding and signaling capacities. In this situation, interventions that target the limiting intracellular protein(s), perhaps in conjunction with a receptor blocker, may be more promising.
Conclusions
Certainly receptor trafficking, as it relates to regulation of both receptor number and compartmentalized signaling, should be a major facet of drug discovery and design. Perhaps more important, however, our appreciation of trafficking may influence the ways in which pharmacological agents are dosed and targeted. In so doing, the sensitivities of various signaling pathways to the number of activated receptors at the cell surface and in endosomes should be considered. Through a combination of dedicated molecular biology, cell biochemistry, subcellular imaging, and mathematical modeling approaches, a framework for handling the associated complexities is currently emerging.
Acknowledgments
Research in the author’s laboratory is supported by the National Science Foundation (grant nos. 0111434 and 0133594), the Whitaker Foundation, and the Camille & Henry Dreyfus Foundation. I thank Douglas Lauffenburger and Alan Wells for their insightful comments on this manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Jason M. Haugh, PhD, is an Assistant Professor of Chemical Engineering and a faculty member of the Genomic Sciences Program at North Carolina State University. E-mail jason_haugh{at}ncsu.edu

