|
|
PAGES DU PRATICIEN
Thérapies ciblant le récepteur du facteur de croissance épidermique dans les cancers aéro-digestifs
Michalis V. Karamouzis, MD;
Jennifer R. Grandis, MD;
Athanassios Argiris, MD
RÉSUMÉ
| |
Contexte Les tumeurs malignes qui se développent à
partir de l'épithélium des voies aéro-digestives,
notamment les cancers bronchiques, de la tête et du cou, et de
l'œsophage, sont les premières causes de mortalité
liée au cancer dans le monde. Compte tenu de l'importance biologique du
récepteur du facteur de croissance épidermique (EGFR) dans le
développement et la progression de ces maladies, les inhibiteurs d'EGFR
se présentent comme de nouvelles thérapies prometteuses.
Objectifs Analyser le statut actuel des inhibiteurs d'EGFR dans les
cancers des voies aéro-digestives supérieures (VADS),
dégager les études en cours visant à optimiser leur
efficacité thérapeutique, et examiner le rôle futur de ces
molécules.
Acquisition des données Recherche systématique sur
MEDLINE des articles de langue anglaise (1966-avril 2007), à l'aide des
termes de recherche EGFR, inhibiteurs d'EGFR, anticorps monoclonaux,
inhibiteurs de tyrosine kinase, cancer bronchique, cancer de la tête et
du cou, cancer œsophagien, et facteurs prédictifs de l'EGFR.
L'évaluation qualitative des études sélectionnées
portait sur la pertinence clinique, avec une insistance particulière
sur le schéma des études contrôlées, la publication
dans des revues à comité scientifique, le nombre adéquat
de patients inclus, l'objectivité des évaluations, et les
techniques utilisées pour minimiser les biais.
Synthèse des données Le rôle de l'EGFR dans la
pathogenèse des cancers des VADS a été largement
étudié, et de multiples stratégies d'inhibition de ce
récepteur sont en cours d'évaluation. L'erlotinib, un inhibiteur
de la tyrosine kinase de l'EGFR utilisé en monothérapie, et le
cetuximab, un anticorps monoclonal anti-EGFR associé à
l'irradiation, ont démontré un bénéfice en termes
de survie dans une étude de patients avec cancer du poumon non à
petites cellules avancé (survie médiane, 6,7 vs 4,7 mois ;
hazard ratio, 0,70 ; intervalle de confiance à 95 %, 0,58-0,87 ; p <
0,001) et dans une étude de patients avec carcinome
épidermoïde de la tête et du cou localement avancé
(survie médiane, 49 vs 29,3 mois ; hazard ratio, 0,74 ; intervalle de
confiance à 95%, 0,57-0,97 ; p = 0,03). Cependant, d'autres
études n'ont pas démontré ces degrés
d'amélioration. Les toxicités des inhibiteurs de l'EGFR incluent
le rash, les diarrhées, et l'hypomagnésémie. Les
mutations somatiques et autres caractéristiques tumorales
moléculaires constituent autant de possibilités
d'individualisation des traitements et de sélection optimale des
patients candidats au traitement anti-EGFR.
Conclusions L'EGFR est une cible thérapeutique prometteuse
dans le traitement des cancers des VADS. De nouvelles recherches
translationnelles sont cependant nécessaires pour optimiser les voies
d'inhibition de ce récepteur, par monothérapies ou associations
thérapeutiques, et pour identifier les patients les plus susceptibles
de bénéficier de ces traitements.
JAMA.
2007;298(1):70-82
Les cancers qui trouvent leur origine dans
l'épithélium des voies aéro-digestives, notamment les
cancers de la tête et du cou, du poumon, et de l'œsophage
(appelés cancers des voies aéro-digestives supérieures
[VADS]), sont les premières causes de mortalité due au cancer
dans le monde, et sont responsables d'environ 2 millions de
décès chaque
année.1
Leur pathogenèse est causalement liée à l'exposition
à des carcinogènes, principalement le tabac et l'alcool ; ils
sont généralement diagnostiqués à des stades
avancés, pour lesquels les traitements disponibles ont un potentiel
curatif limité. Des avancées même infimes dans la prise en
charge des cancers des VADS peuvent avoir des répercussions sur des
millions de vies. La pierre angulaire de l'ère thérapeutique
actuelle réside dans le développement rationnel
d'anticancéreux, qui a été favorisé par la
connaissance approfondie de la biologie du cancer. Les caractéristiques
séminales du cancer relatives à la croissance cellulaire, au
potentiel de réplication, aux voies de signalisation, aux interactions
entre cellules, et à l'angiogenèse entre autres, sont
progressivement dévoilées et avantageusement exploitées
à des fins thérapeutiques, ce qui permet un développement
accéléré des nouveaux composés.
La carcinogenèse épithéliale est le résultat
phénotypique d'altérations génétiques et
épigénétiques successives qui entraînent la
dérégulation de l'homéostasie
cellulaire.2
Les données cumulées suggèrent que les voies de
signalisation des récepteurs membranaires pourraient contribuer
à l'acquisition du phénotype malin. Le récepteur du
facteur de croissance épidermique (EGFR) est un récepteur
membranaire à tyrosine kinase (TK) de type I, qui régule des
fonctions cellulaires clés dans les tumeurs malignes
épithéliales.3
Plusieurs études utilisant des méthodes diverses ont
démontré que l'EGFR est fréquemment surexprimé
dans les cancers des VADS, typiquement plus dans les carcinomes
épidermoïdes que dans les adénocarcinomes. L'expression
protéique de l'EGFR est détectée dans environ 50 % ou
plus des cancers bronchiques non à petites cellules
(CBNPC),4-7
dans plus de 90 % des carcinomes épidermoïdes de la tête et
du cou
(HNSCC),8 et
dans 30 % à 70 % des cancers
œsophagiens.9-11
Des niveaux élevés d'expression protéique de l'EGFR ont
été corrélés à une moins bonne survie des
patients dans les CBNPC, bien que l'association n'ait pas été
robuste et les résultats disparates en fonction des
études,12-18
des
HNSCC,8,19
et des cancers
œsophagiens.10,11,12
En outre, le nombre élevé de copies du gène EGFR, qui est
variablement corrélé à l'expression protéique de
l'EGFR, a également été rapporté comme un marqueur
de pronostic péjoratif dans les cancers des
VADS.5,21
En conséquence, l'EGFR représente une cible rationnelle dans le
traitement de ces cancers. Bien que le blocage de la fonction de l'EGFR puisse
être obtenu par diverses méthodes
(Tableau 1), 2 approches ont
été plus largement étudiées en laboratoire et en
clinique: les anticorps monoclonaux (AcMo) dirigés contre le domaine
extracellulaire du récepteur et les petites molécules inhibant
la TK du domaine intracellulaire de l'EGFR (TKI-EGFR). Certaines
molécules de ces deux catégories ont été
intégrées dans la prise en charge standard des cancers des VADS.
Cependant, leur application clinique a engendré un certain nombre de
problèmes.22
L'identification des facteurs moléculaires pronostiques potentiellement
susceptibles d'optimiser la sélection des patients et
l'efficacité thérapeutique continue de faire l'objet d'une
intense recherche fondamentale et clinique. Dans le présent article,
nous résumerons l'état actuel des évaluations cliniques
sur les inhibiteurs d'EGFR dans les cancers des VADS, dégagerons les
études en cours relatives aux facteurs moléculaires
prédictifs, et examinerons les futures perspectives de ces agents.
|
|
|
|
Tableau 1.. Stratégies thérapeutiques ciblant le récepteur du
facteur de croissance épidermique (EGFR)
|
|
|
ACQUISITION DES DONNÉES
Nous avons effectué, sur MEDLINE, une recherche dans les articles de
langue anglaise (de 1966 à avril 2007) en utilisant les termes EGFR,
inhibiteurs d'EGFR, anticorps monoclonaux, inhibiteurs de tyrosine kinase,
cancer bronchique, cancer de la tête et du cou, cancer œsophagien,
et facteurs prédictifs de l'EGFR. Les bibliographies pertinentes de la
littérature ont été revues manuellement pour
compléter les résultats de la recherche. Pour
l'évaluation des bénéfices des inhibiteurs d'EGFR dans
les cancers des VADS, les données des études randomisées
ont été privilégiées. L'évaluation
qualitative des études sélectionnées incluait en outre la
publication dans des revues à comité scientifique, un nombre
adéquat de patients inclus, l'objectivité des
évaluations, et les techniques utilisées pour minimiser les
biais. Lors de la revue des études, les critères cliniques
d'intérêt, par ordre décroissant d'importance, incluaient
le bénéfice en termes de survie, la survie sans progression, le
taux de réponse, le taux de stabilisation de la maladie, et
l'amélioration de la qualité de vie. Des informations
complémentaires ont été obtenues dans les
présentations orales et les résumés des congrès
2004-2006 de l'ASCO (American Society of Clinical Oncology) et 2004-2007 de
l'AACR (American Association for Cancer Research). Les recommandations
publiées du NNCN (National Comprehensive Cancer Network) et de l'ASCO
ont également été revues. Les informations sur les
études cliniques en cours ont été obtenues sur le site
Internet des NIH américains (US National Institutes of Health)
(http://www.clinicaltrials.gov).
SYNTHÈSE DES DONNÉES
Biologie de l'EGFR
L'EGFR (erbB-1) est un membre de la famille des récepteur tyrosine
kinase erbB, qui inclut également erbB-2, erbB-3 et erbB-4. Ces
récepteurs possèdent un domaine de liaison au ligand
extracellulaire, un domaine d'ancrage transmembranaire, et un domaine
intracellulaire à activité tyrosine kinase -à l'exception
de erbB-3. La liaison d'un ligand comme l'EGF (facteur de croissance
épidermique) ou le TGF- (transforming growth factor )
à l'EGFR entraîne sa dimérisation avec un autre EGFR ou
avec un autre membre de la famille
erbB.23 La
dimérisation de l'EGFR provoque l'activation de la TK associée
au récepteur, le recrutement de complexes de signalisation, et la
phosphorylation (activation) de multiples cascades en
aval.24 Les
réseaux activés de l'EGFR incluent les voies de
Ras/MAPKinase,25
de la phospatidylinositol
3-kinase/AKT,26
de
JAK/STAT,27
et de la protéine kinase C/phospholipase-C (transforming growth factor )
à l'EGFR entraîne sa dimérisation avec un autre EGFR ou
avec un autre membre de la famille
erbB.23 La
dimérisation de l'EGFR provoque l'activation de la TK associée
au récepteur, le recrutement de complexes de signalisation, et la
phosphorylation (activation) de multiples cascades en
aval.24 Les
réseaux activés de l'EGFR incluent les voies de
Ras/MAPKinase,25
de la phospatidylinositol
3-kinase/AKT,26
de
JAK/STAT,27
et de la protéine kinase C/phospholipase-C
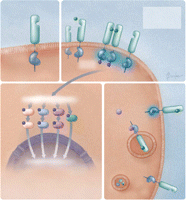
 28
(Figure). Ces cascades sont des
régulateurs puissants de divers processus intracellulaires et
intercellulaires comme la prolifération, l'apoptose, l'invasion,
l'angiogenèse, et l'envahissement métastatique. L'activation de
l'EGFR peut également être obtenue par interaction avec d'autres
récepteurs, comme les récepteurs couplés aux
protéines
G,29 les
récepteurs au PDGF (platelet-derived growth
factor),30
le récepteur de l'IGF-1 (insulin-like growth
factor),31
et les récepteurs
hormonaux32
(Figure). La régulation
de l'EGFR à la surface cellulaire comporte une composante essentielle
qui consiste en un processus d'endocytose en 2
étapes.33
Tout d'abord, une internalisation rapide induite par le ligand retire les
récepteurs activés de la membrane cellulaire et les enclave dans
les endosomes. Ensuite, les récepteurs internalisés sont soit
dirigés vers les lysosomes où ils subissent une
dégradation, soit recyclés vers la membrane plasmique. La
sélection de l'EGFR pour la dégradation lysosomiale
nécessite une autophosphorylation du récepteur, qui recrute
ensuite la Cbl - une ubiquitine E3-ligase responsable de l'ubiquitination de
l'EGFR24
(Figure). L'endocytose du
récepteur induite par le ligand produit généralement une
régulation négative de la signalisation de l'EGFR, même si
certaines données suggèrent que les récepteurs
internalisés pourraient conserver leur capacité de couplage aux
protéines effectrices et d'activation des cascades de
signalisation.34 28
(Figure). Ces cascades sont des
régulateurs puissants de divers processus intracellulaires et
intercellulaires comme la prolifération, l'apoptose, l'invasion,
l'angiogenèse, et l'envahissement métastatique. L'activation de
l'EGFR peut également être obtenue par interaction avec d'autres
récepteurs, comme les récepteurs couplés aux
protéines
G,29 les
récepteurs au PDGF (platelet-derived growth
factor),30
le récepteur de l'IGF-1 (insulin-like growth
factor),31
et les récepteurs
hormonaux32
(Figure). La régulation
de l'EGFR à la surface cellulaire comporte une composante essentielle
qui consiste en un processus d'endocytose en 2
étapes.33
Tout d'abord, une internalisation rapide induite par le ligand retire les
récepteurs activés de la membrane cellulaire et les enclave dans
les endosomes. Ensuite, les récepteurs internalisés sont soit
dirigés vers les lysosomes où ils subissent une
dégradation, soit recyclés vers la membrane plasmique. La
sélection de l'EGFR pour la dégradation lysosomiale
nécessite une autophosphorylation du récepteur, qui recrute
ensuite la Cbl - une ubiquitine E3-ligase responsable de l'ubiquitination de
l'EGFR24
(Figure). L'endocytose du
récepteur induite par le ligand produit généralement une
régulation négative de la signalisation de l'EGFR, même si
certaines données suggèrent que les récepteurs
internalisés pourraient conserver leur capacité de couplage aux
protéines effectrices et d'activation des cascades de
signalisation.34
|
|
|
|
Figure.. Activation, traitement et signalisation du récepteur du facteur de
croissance épidermique
ATP indique l'adénosine triphosphate; Cbl, une ubiquitine E3-ligase;
IGF, facteur de croissance analogue à l'insuline; JAK, Janus kinase;
MAPK, mitogen-activated protein kinase; P, phosphate; PDGF, platelet-derived
growth factor; PI3K, phosphatidylinositol-3-kinase; PKC, protéine
kinase C; PLC-, phospholipase C; STAT, signal transducer and
activator of transcription; TK, tyrosine kinase; Ub, ubiquitine.
|
|
|
Les manifestations pathologiques de l'EGFR dans les cancers des VADS
résultent d'au moins 4 mécanismes majeurs: (1) la surexpression
des ligands de l'EGFR et la formation de boucles autocrine/paracrine, (2)
l'activation mutationnelle de l'EGFR, (3) l'amplification de l'EGFR, et (4) la
transactivation par d'autres tyrosines kinases de type récepteur et
non-récepteur.23,35 Deux types majeurs de mutations d'EGFR ont
été identifiés : la délétion du domaine
extracellulaire et les mutations somatiques dans l'activité TK du
domaine intracellulaire. Plusieurs classes de mutations de l'ectodomaine
d'EGFR (vI-vVII) ont été découvertes, bien que l'EGFRvIII
soit la plus fréquemment trouvée dans les cancers des VADS. Il a
récemment été montré que le mutant EGFRvIII
présent dans les HNSCC induisait une activation constitutive du
récepteur indépendante du ligand, un renforcement des effets en
aval, et la résistance au ciblage de l'EGFR de type
sauvage.36
Les mutations activatrices du domaine TK de l'EGFR sont plus
fréquemment observées dans les CBNPC que dans les autres cancers
des
VADS.37-40
Les mutations les plus fréquentes de l'EGFR dans les CBNPC sont des
délétions dans l'exon 19 et des substitutions de
nucléotides dans l'exon 21. Ces mutations affectent la poche de liaison
à l'ATP (adénosine triphosphate) du domaine TK de l'EGFR et
confèrent au récepteur une activité TK non
régulable. Les tumeurs présentant des mutations d'EGFR
répondent mieux aux TKI-EGFR, ce qui suggère qu'elles
dépendent de la signalisation de l'EGFR mutant. Il convient cependant
de noter qu'en dépit de l'importance des anomalies des voies
moléculaires induites par EGFR, elles ne peuvent pas être
considérées comme la seule altération
génétique dominante dans la pathogenèse
moléculaire des cancers des
VADS.41
La signalisation de l'EGFR est essentielle dans divers tissus normaux,
notamment la peau, l'intestin et les reins, ce qui explique le
développement de certaines toxicités résultant du ciblage
de ce récepteur, comme les rash, les diarrhées, et
l'hypomagnésémie. Un rash acnéiforme et d'autres
toxicités cutanées ont ainsi été observés
chez la majorité des patients recevant des inhibiteurs
d'EGFR.42 En
outre, le blocage de l'EGFR peut interférer avec l'absorption de
magnésium, possiblement en raison de la forte expression de l'EGFR dans
la branche ascendante de l'anse de Henle, où la plus grande partie du
magnésium filtré est réabsorbée ; ceci pourrait
expliquer la fréquence élevée
d'hypomagnésémie observée au cours du traitement par
cetuximab.43,44
Stratégies d'inhibition de l'EGFR
Diverses stratégies ont été développées
pour interrompre la voie de transduction du signal EGFR
(Tableau 1). Parmi elles, les
anticorps monoclonaux (AcMo) anti-EGFR et les TKI-EGFR ont fait l'objet des
recherches les plus approfondies. Les mécanismes putatifs de
l'activité anticancéreuse basée sur les AcMo anti-EGFR
peuvent être classés en deux catégories. Tout d'abord, ils
empêchent la liaison des ligands au domaine extracellulaire de l'EGFR,
inhibent la dimérisation/activation subséquente du
récepteur, et induisent sa
dégradation.45
Le second mécanisme potentiel du traitement par AcMo anti-EGFR est une
action indirecte médiée par le système immunitaire. Il a
été observé que certains mécanismes effecteurs
immuns spécifiques étaient déclenchés par les
anticorps monoclonaux thérapeutiques, notamment la cytotoxicité
cellulaire dépendant des anticorps, la cytotoxicité
complément-dépendante, et la cytotoxicité à
médiation cellulaire dépendant du
complément.46,47
Un mode alternatif de ciblage de l'EGFR consiste à associer les
anticorps monoclonaux à des toxines pour combattre sélectivement
les cellules tumorales surexprimant
EGFR.48
Les tyrosines kinases de type récepteur et non récepteur sont
des médiateurs clés dans les voies de signalisation de l'EGFR,
et beaucoup sont dérégulées au cours de la
tumorigenèse dans les VADS. Les petites molécules inhibitrices
qui ciblent ces TK agissent directement dans les cellules tumorales au lieu de
médier les réponses immunitaires. La plupart des petites
molécules inhibitrices de TK sont des agonistes de l'adénosine
triphosphate, et rivalisent ainsi (de manière réversible ou
irréversible) avec la liaison de l'ATP dans la fente catalytique de la
kinase. À la différence des anticorps monoclonaux, elles passent
la membrane plasmatique et interagissent avec le domaine cytoplasmique des
récepteurs à la surface des cellules ou modulent les
molécules de signalisation intracellulaires. Les inhibiteurs de la
tyrosine kinase ont une sélectivité variable pour les TK, et
certains ont une action double ou multicible
(Tableau 1). Les TKI-EGFR sont
généralement supposés être moins spécifiques
que les anticorps monoclonaux ; cette caractéristique est
potentiellement avantageuse pour l'activité anti-tumorale, mais elle
peut être associée à des toxicités majorées,
dues à l'inhibition de plusieurs voies de signalisation susceptibles
d'interférer avec les fonctions cellulaires normales. Un certain nombre
d'inhibiteurs TK de nouvelle génération présentent une
activité à large spectre contre divers TK de type
récepteur (EGFR, récepteur du facteur de croissance vasculaire
endothélial [VEGF]), TK de type non récepteur (kinase Src par
exemple), et/ou molécules en aval
(ras/MAPK).49
Le développement de nouveaux inhibiteurs d'EGFR a également
été entreprise en utilisant des méthodes de triage
à haut débit, une modélisation moléculaire
rationnelle, ou l'association des deux. Le triage à haut débit
des composés constitue un moyen d'identifier de nouveaux inhibiteurs
d'EGFR, qui peuvent ensuite être améliorés par des
approches pharmacochimiques pour développer des médicaments
éventuellement utilisables dans les tumeurs avec résistance
acquise aux anti-EGFR
actuels.50
Des approches structurelles rationnelles ont été
utilisées pour concevoir des molécules réprimant
l'expression de l'acide nucléique, comme par exemple les
oligonucléotides antisens, l'ARN interférence, et les ribozymes,
qui empêchent la traduction de l'ARN messager (ARNm) de l'EGFR ; elles
sont encore en première phase de
développement.51
La technologie antisens utilise une séquence complémentaire
à une cible d'ARNm spécifique (EGFR par exemple), ce qui inhibe
son expression et le transfert de l'information génétique de
l'ADN au niveau protéique. L'ARN interférence a
été récemment reconnu comme un mécanisme puissant
permettant de réprimer l'expression du gène par
dégradation ciblée de l'ARNm. De courtes molécules d'ARN
double brin, appelées petits ARN interférents et conçus
pour être homologues à la séquence d'ARNm de la
protéine cible, peuvent être introduits dans le cytoplasme et
déclencher l'hydrolyse de l'ARNm par le biais de l'ARN
interférence. Des études précliniques utilisant ces
techniques ont démontré une inhibition spécifique de la
croissance des cellules tumorales, ainsi que des effets inhibiteurs
synergiques avec les substances chimiothérapeutiques
conventionnelles.52
Cependant, les traitements ciblant les acides nucléiques
nécessitent des études complémentaires chez les patients
porteurs de cancers des VADS, afin de déterminer leur activité
anti-tumorale, leurs modalités d'adminis tration optimales et leur
toxicité.
Cancer bronchique. Le cancer bronchique est la première cause
de décès par cancer dans le monde ; la plupart des patients
développent une maladie métastatique à distance, dont ils
décèdent.1
Les agents cytotoxiques les plus récents, développés au
cours des dix dernières années, n'ont que marginalement
influé sur la survie. Le développement de nouveaux traitements
systémiques qui pourraient améliorer le pronostic des patients
est donc extrêmement nécessaire. Les avancées
réalisées dans la compréhension de la pathogenèse
moléculaire du cancer du poumon ont permis l'identification de cibles
potentielles pour des interventions thérapeutiques sélectives.
Les agents ciblant l'EGFR ont été les premiers à
être testés avec succès chez des patients avec CBNPC
avancé.53,54
Les TKI-EGFR gefitinib et erlotinib ont obtenu des taux de réponses
de respectivement 10 % et 20 % dans des études cliniques de phase 2,
chez des patients avec CBNPC avancé, précédemment
traité.55-57
Sur la base de ces observations, 2 études de phase 3
randomisées, en double aveugle, contrôlées contre placebo,
ont été initiées pour évaluer la survie globale
sous TKI-EGFR comparé au meilleur traitement d'appoint utilisé
seul chez des patients avec CBNPC avancé, ayant reçu une
chimiothérapie en première ou deuxième ligne
(Tableau 2). Dans la
première étude, les patients étaient randomisés
pour recevoir soit de l'erlotinib (150 mg par voie orale, une fois par jour)
soit un placebo. La survie globale était de 6,7 mois pour les patients
du groupe erlotinib comparé à 4,7 mois chez les patients du
groupe témoin (p <
0,001).58
L'évaluation de la qualité de vie a révélé
que les patients du groupe erlotinib bénéficiaient d'un
délai plus long jusqu'à la dégradation des
symptômes associés à la
tumeur.68
|
|
|
|
Tableau 2.. Études cliniques de phase 3 achevées utilisant des anti-EGFR
dans les cancers des voies aéro-digestives
|
|
|
Dans la seconde étude, le gefitinib à la dose quotidienne de
250 mg n'améliorait pas la survie globale comparé au placebo,
dans une cohorte similaire de patients avec CBNPC, même si une analyse
en sous-groupe démontrait un bénéfice en survie chez les
Asiatiques et les
non-fumeurs.61
Les raisons expliquant les divergences de résultats de ces 2
études cliniques au schéma similaire ne sont pas claires. La
possibilité que le gefitinib ait été administré
à une dose biologique suboptimale a été proposée
comme une explication plausible ; elle est en outre soutenue par des
observations
précliniques.69
La question de savoir si l'escalade de dose du gefitinib au-delà de 500
mg améliorerait son efficacité clinique reste à
démontrer dans des
études.70
Le gefitinib a également produit des résultats négatifs
en monothérapie adjuvante, après traitement par modalités
combinées de chimiothérapie et radiothérapie, dans une
étude de phase 3 chez des patients avec CBNPC de stade
III64
(Tableau 2). Bien qu'une
étude de phase 3 sur le gefitinib adjuvant, incluant des patients avec
CBNPC réséqué, ait été
prématurément interrompue, l'utilisation de l'erlotinib en
traitement adjuvant ou d'entretien est actuellement en cours
d'évaluation (Tableau
3).
|
|
|
|
Tableau 3.. Études cliniques de phase 3 en cours sélectionnées
utilisant des anti-EGFR dans les cancers des voies aéro-digestives
|
|
|
Quatre études de phase 3 ont évalué un traitement par
TKI-EGFR, erlotinib ou gefitinib, associé à une
chimiothérapie standard de première ligne chez des patients avec
CBNPC
avancé59,60,62,63
(Tableau 2). Aucun
bénéfice en termes de survie ou de tout autre critère
d'efficacité n'a pu être mis en évidence avec l'adjonction
d'un TKI-EGFR à la chimiothérapie dans ces études,
malgré une étude préclinique sur des greffes
hétérologues de tumeur humaine en faveur de ces
associations.53
Il a été postulé que les TKI-EGFR et la
chimiothérapie seraient plus efficaces en administration
séquentielle plutôt que simultanée, et ce concept est en
cours d'évaluation
clinique71
(Tableau 3). Cependant, il est
probable que l'absence de bénéfice observée dans les
études de phase 3 évaluant les inhibiteurs d'EGFR
associés à la chimiothérapie ait été due au
manque de sélection des patients, à la stratification
basée sur les caractéristiques moléculaires des tumeurs,
ou aux deux. Ainsi, le plan de ces études ne prenait pas en compte la
présence de mutations activant l'EGFR, le nombre de copies du
gène EGFR, voire les deux, qui sont récemment apparus comme des
facteurs prédictifs de l'efficacité des TKI-EGFR. En
conséquence, des bénéfices modestes peuvent ne pas avoir
été détectés en raison de
l'hétérogénéité moléculaire de la
population étudiée.
Les anticorps anti-EGFR, principalement le cetuximab, ont également
été étudiés dans le CBNPC, seuls et en association
avec la chimiothérapie. Dans une étude de 66 patients avec CBNPC
avancé, précédemment traité, le cetuximab en
monothérapie produisait un faible taux de réponse (4,5 %), mais
une survie globale médiane prometteuse de 8,9
mois.72 Un
certain nombre d'études randomisées de phase 3 en cours
évaluent l'adjonction du cetuximab à la chimiothérapie
dans le traitement de première et deuxième ligne du CBNPC
avancé (Tableau 3).
Cancers de la tête et du cou. L'EGFR a un rôle majeur
dans la pathogenèse des HNSCC, et sa surexpression a été
rapportée dans plus de 80 % des
cas.23 En
outre, la régulation positive de l'EGFR a également
été documentée dans l'épithélium
apparemment normal adjacent au tissu malin, soutenant la notion de «
champ de cancérisation » qui se réfère au
développement multifocal des lésions
précancéreuses et malignes dans l'ensemble de
l'épithélium exposé aux
carcinogènes.73
En conséquence, l'inhibition de la signalisation de l'EGFR
représente une nouvelle stratégie rationnelle dans le traitement
des HNSCC.
Plusieurs inhibiteurs de TK avec une activité préclinique
contre EGFR ont été évalués dans les HNSCC. De
nombreuses études de phase 2 avec un seul groupe, utilisant le
gefitinib ou l'erlotinib, ont montré une activité modeste en
monothérapie (taux de réponses de 1 %-11 %) chez des patients
avec HNSCC récidivant ou
métastatiques;74-77
dans ce même contexte, le lapatinib, un double inhibiteur anti-EGFR et
ERBB2 récent, n'a pas démontré
d'efficacité.78
Récemment, des résultats préliminaires prometteurs issus
d'études cliniques évaluant l'association du gefitinib ou de
l'erlotinib avec des agents chimiothérapiques, ou d'autres agents
biologiques (comme le bevacizumab) avec une radiothérapie, ont
été
rapportés.79
En outre, 2 études de phase 3 ont évalué le gefitinib
dans des HNSCC en récidive ou métastatiques.
La première était une étude multicentrique
internationale qui comparait le gefitinib (à la dose quotidienne de 250
mg ou de 500 mg) au méthotrexate ; elle a récemment
rapporté des résultats démontrant que le gefitinib n'est
pas supérieur au méthotrexate dans ce
contexte65
(Tableau 2) ; la seconde est
une étude en cours qui compare le docétaxel avec ou sans
gefitinib, en première ou deuxième ligne de
traitement79
(Tableau 3).
Le cetuximab a été le premier de ces agents à obtenir
l'AMM dans le traitement des patients avec HNSCC aux États-Unis. Cette
approbation était basée sur des données de survie
positives, issues d'une étude clinique randomisée de phase 3 qui
comparait l'irradiation et le cetuximab à l'irradiation seule chez des
patients avec HNSCC localement
avancé67
(Tableau 2).
L'amélioration du contrôle locorégional et de la survie
obtenue avec l'adjonction du cetuximab à l'irradiation était
comparable à celle obtenue avec des traitements à base de
platine. Cependant, aucune étude randomisée n'a encore
comparé l'irradiation plus cetuximab à l'irradiation plus
cisplatine. Les recherches se sont préférentiellement
axées sur un nouveau renforcement thérapeutique par adjonction
du cetuximab à la chimioradiothérapie à base de platine.
Plusieurs études de phase 2, achevées ou actuellement en cours,
ont évalué des associations de l'irradiation avec le cetuximab
et des dérivés de platine. Une étude de phase 2
récemment rapportée évaluait l'efficacité et la
toxicité de l'association du cetuximab avec le cisplatine et la
radiothérapie hyperfractionnée et
accélérée, avec des résultats d'efficacité
préliminaires prometteurs. Cependant, cette étude a
été prématurément interrompue en raison
d'événements indésirables significatifs, dont 2
décès qui peuvent ne pas avoir été
spécifiquement liés au cetuximab.80 Une étude de phase 3
compare actuellement cette association à la radiothérapie et au
cisplatine seuls (Tableau
3).
Le cetuximab a également été largement
étudié dans le traitement des HNSCC en récidive ou
métastatiques. Le groupe ECOG (Eastern Cooperative Oncology Group) a
mené une étude randomisée qui comparait le cisplatine
avec ou sans cetuximab dans le traitement de première intention de
HNSCC récidivants ou métastatiques.66 Bien que cette
étude ait présenté une petite taille
d'échantillon, avec 117 patients analysables, elle démontrait
une amélioration significative des taux de réponse et une
tendance en faveur d'une amélioration de la médiane de survie
sans progression avec l'adjonction du cetuximab
(Tableau 2). Le cetuximab a
également démontré une efficacité chez des
patients avec HNSCC réfractaire au platine, en
monothérapie81
-ce qui lui a permis d'obtenir l'AMM également dans cette indication
aux États-Unis- ou en association avec le
platine.82,83
Une étude de phase 3 récemment achevée a
évalué l'association d'un dérivé du platine
(cisplatine ou carboplatine) et du 5-fluorouracile avec ou sans cetuximab,
dans des HNSCC récidivants ou
métastatiques84
(Tableau 3).
Cancer œsophagien. Le carcinome épidermoïde de
l'œsophage reste une maladie
fréquente.1
Cependant, l'incidence de l'adénocarcinome de l'œsophage augmente
graduellement. Ces 2 entités peuvent être distinctes dans leur
étiologie, leur pathogenèse et leur
pronostic.85
Plusieurs études ont rapporté une expression protéique de
l'EGFR de 30 % à 70 % dans les carcinomes œsophagiens; elle est
plus fréquente dans les carcinomes épidermoïdes que dans
les
adénocarcinomes9-11
et a été corrélée à un pronostic
péjoratif, ainsi qu'à une réponse inférieure au
traitement conventionnel. La surexpression de l'EGFR a été
documentée dans environ 15 % des carcinomes
œsophagiens,9,86
tandis que les mutations d'EGFR y sont rarement observées (0 %-11
%).9,39,87,88
Des études de phase 2 utilisant des TKI-EGFR en monothérapie, en
première et deuxième ligne de traitement chez des patients non
sélectionnés avec un cancer œsophagien avancé, ont
démontré une activité modeste ;86,89 l'association
d'inhibiteurs de la tyrosine kinase ou d'anticorps monoclonaux avec la
chimiothérapie, la radiothérapie, ou les deux, est actuellement
en cours
d'évaluation.90,91
Marqueurs prédictifs d'efficacité du traitement anti-EGFR
Facteurs prédictifs cliniques. Des études cliniques, portant
sur des TKI-EGFR chez des patients avec BNPC, ont identifié un certain
nombre de caractéristiques prédictives d'un
bénéfice majoré des TKI-EGFR, telles que l'absence
d'antécédent de tabagisme, le sexe féminin, l'histologie
d'adénocarcinome ou de carcinome bronchiolo-alvéolaire (CBA), et
l'origine
asiatique.54
La base moléculaire de ces associations n'est pas toujours
évidente. En outre, la distinction entre les facteurs pronostiques et
prédictifs peut être potentiellement établie que dans des
études randomisées incluant un groupe de traitement sans
inhibiteur
d'EGFR.92 La
plus grande probabilité de réponse aux TKI-EGFR chez les
patients sans antécédent de tabagisme avec CBNPC est une
observation constante et particulièrement fascinante, qui pourrait
être partiellement liée à la fréquence plus
élevée de mutations de l'EGFR dans ce
groupe.93 En
outre, les patients avec un CBA pur ou un adénocarcinome à type
de CBA semblent tirer un bénéfice substantiel des TKI-EGFR,
comme cela a été démontré dans des essais de phase
2,94,95
bien qu'aucune donnée corroborante d'études de phase 3 ne soit
encore disponible.
Il a également été démontré de
manière constante que l'apparition et la sévérité
du rash cutané dû aux TKI-EGFR et aux anticorps monoclonaux sont
corrélées à l'évolution du patient dans de
nombreux types de tumeurs, incluant les HNSCC et les
CBNPC.96 Les
toxicités cutanées associées aux inhibiteurs d'EGFR sont
attribuables au rôle prédominant de l'EGFR dans la physiologie
cutanée.42
Une hypothèse intéressante est que les patients porteurs de
certains polymorphismes dans l'intron 1 du gène EGFR pourraient
présenter une plus grande probabilité de réponse tumorale
favorable et de développement de s cutanées avec un traitement
inhibiteur
d'EGFR.97
Marqueurs prédictifs moléculaires. Mutations de
l'EGFR. L'activation de mutations somatiques dans le domaine TK de l'EGFR a
été décrite pour la première fois dans les
CBNPC.37,38
Il convient de noter que ces mutations sont présentes chez une partie
des patients avec CBNPC, qui varie en fonction de la race ou
l'ethnicité (environ 10 % dans des populations non
sélectionnées aux États-Unis, mais proportion
significativement supérieure dans le Sud-Est asiatique), de l'histoire
de tabagisme, du sexe et de
l'histologie.98,99
Des mutations similaires ont été détectées dans
les HNSCC, mais moins fréquemment (1 %-7
%),40,100
ainsi que dans les carcinomes œsophagiens (0 %-11
%).9,39,87,88
La présence de mutations activatrices de l'EGFR semble être un
facteur pronostique associé à une survie prolongée chez
les patients avec CBNPC avancé, traité par
chimiothérapie, avec ou sans
TKI-EGFR.92
En outre, dans de multiples études des
CBNPC,101-103
mais pas dans une étude randomisée, contrôlée
contre placebo, sur les CBNPC avancés traités par
erlotinib,104
les mutations de l'EGFR étaient prédictives de survie chez les
patients traités par TKI-EGFR. Une mutation ponctuelle secondaire dans
le domaine TK de l'EGFR, T790M, qui est associée à une
résistance acquise, a également été
rapportée.105
Bien que les mutations de l'EGFR soient des facteurs pronostiques majeurs
potentiels, elles ne peuvent que partiellement expliquer le
bénéfice clinique documenté des TKI-EGFR chez les
patients avec CBNPC, alors que dans d'autres cancers des VADS, ces mutations
se révèlent de signification
limitée.106
Plusieurs études cliniques évaluent actuellement les TKI-EGFR en
première ligne de traitement chez des patients naïfs de
chimiothérapie avec CBNPC avancé et mutations activatrices de
l'EGFR.107-109
Jusqu'à présent, tant le gefitinib que l'erlotinib ont
démontré une activité anti-tumorale spectaculaire, avec
des taux de réponses objectives de 75 % à 82 % dans ce
contexte.107-109
Une limite potentielle à l'évaluation prospective des
mutations de l'EGFR réside dans la difficulté à obtenir
suffisamment de tissu tumoral pour l'analyse, chez les patients avec un CBNPC
inopérable. La détection des mutations dans l'épanchement
pleural malin est en cours
d'étude.110
Nombre de copies et polymorphismes du gène EGFR. Le nombre de copies du
gène EGFR est apparu comme un facteur prédictif majeur de
l'efficacité des inhibiteurs d'EGFR dans les CBNPC. Un nombre
élevé de copies du gène, une amplification ou une
polysomie élevée, évalués par hybridation in situ
fluorescente, ont été documentés dans 31 % à 45 %
des cas de CBNPC, et associés à une réponse
majorée au traitement par TKI-EGFR, ainsi qu'à une survie plus
longue.104,111-113
Le rôle potentiel de l'amplification des gènes de l'EGFR en
tant que facteur pronostique est également en cours d'étude dans
les HNSCC21
et dans les carcinomes épidermoïdes de l'œsophage;9 des
données relatives à son utilisation comme facteur pronostique
dans des études sur les inhibiteurs d'EGFR dans ces cancers sont
attendues. La signification de l'amplification des gènes pour les
autres membres de la famille erbB (notamment ErbB-2, ErbB-3) chez les patients
traités par inhibiteurs d'EGFR est également en cours
d'évaluation.114,115
La régulation de l'expression de l'EGFR complexe et implique plusieurs
étapes. Les variations polymorphiques ethniques ou interindividuelles
du gène EGFR peuvent également altérer l'expression du
récepteur à l'EGF, son activité, ou les
deux.116,117
Ces variations pourraient en outre être responsables des
différences observées dans l'efficacité du traitement
anti-EGFR et devront être mieux
étudiées.118
Récemment, un modèle pharmacocinétique a
été développé pour évaluer l'influence de
l'activité du cytochrome P450 3A sur le métabolisme des
TKI-EGFR, leur activité anti-tumorale, et leur
toxicité.119
Expression protéique de l'EGFR et autres marqueurs
moléculaires. Un niveau d'expression protéique
élevé de l'EGFR, déterminé par immunohistochimie,
est observé dans la grande majorité des cancers des VADS, le
niveau d'expression le plus élevé étant observé
dans les
HNSCC.23,120
Cependant, aucune donnée n'a suggéré que l'expression
protéique de l'EGFR était uniformément
corrélée à l'activité anti-tumorale des
inhibiteurs
d'EGFR.66,81,104,113,120,121
Ainsi, des résultats variables ont été obtenus dans des
études évaluant l'expression protéique de l'EGFR et
l'activité des TKI-EGFR dans les CBNPC
avancés.122
La divergence des résultats concernant la valeur prédictive de
l'expression protéique de l'EGFR pourrait être partiellement
attribuée à la variabilité dans les
caractéristiques de la population et dans la méthodologie
employée. Néanmoins, l'analyse d'études de phase 3
suggère que les patients avec CBNPC négatif pour l'expression
protéique de l'EGFR n'obtiennent aucun bénéfice en termes
de survie avec les
TKI-EGFR.104,113
Des perturbations dans les voies de ras/MAPK, PI3K-3/AKT, et JAK/STAT, qui
sont les principaux effecteurs en aval de la signalisation de l'EGFR, sont en
cours d'étude en tant que facteurs pronostiques
potentiels.123,124
Il a ainsi été récemment démontré in vitro
que la phosphorylation d'AKT sans stimulation du ligand de l'EGFR avait un
rôle majeur dans la sensibilité aux
TKI-EGFR,125
probablement dû à l'activation médiée par
erbB-3.126
Le CBNPC des fumeurs semble présenter un spectre différent
d'anomalies moléculaires comparé au CBNPC des non-fumeurs, ce
qui suggère des différences survenant précocement dans la
carcinogenèse et potentiellement associées au
pronostic.127
Les mutations K-ras sont fortement corrélées à l'histoire
de tabagisme et ont été associées à un pronostic
péjoratif.128
Les mutations activatrices de K-ras et de l'EGFR pourraient être
mutuellement exclusives, dans la mesure où elles ne surviennent
pratiquement jamais dans la même
tumeur.129
Il apparaît que la croissance tumorale peut dépendre de
l'activation de l'une ou l'autre voie, mais pas des deux. L'ensemble des
données précliniques et cliniques suggère que les
patients avec CBNPC et mutations de K-ras pourraient représenter une
population distincte avec une résistance intrinsèque aux
stratégies thérapeutiques
anti-EGFR.92,130,131
En dépit de l'accroissement rapide des données sur les
facteurs prédictifs d'efficacité, aucun biomarqueur ne peut
à ce jour être recommandé pour sélectionner les
patients candidats aux traitements anti-EGFR. Il est possible qu'un algorithme
intégrant un panel de biomarqueurs, incluant les mutations de l'EGFR,
le nombre de copies du gène EGFR ou les taux protéiques d'EGFR,
puisse constituer un outil clinique utile.
Perspectives futures
L'introduction des inhibiteurs d'EGFR a constitué l'une des
premières étapes du développement des thérapies
ciblées réussies dans les cancers des VADS. Optimiser les
stratégies et tirer les leçons des échecs constituent
sans aucun doute des paliers essentiels dans le processus de
développement de ces agents. Les événements
moléculaires impliqués dans la résistance
intrinsèque ou acquise au traitement anti-EGFR restent encore à
dévoiler entièrement. L'une des stratégies potentielles
pour l'amélioration des effets anti-tumoraux est le traitement
combiné avec des agents anti-EGFR distincts, susceptibles de maximiser
l'inhibition de la signalisation de l'EGFR.132 En conséquence,
l'association des TKI-EGFR et des anticorps monoclonaux est
intéressante et est évaluée en étude clinique.133
Cependant, le traitement anti-EGFR seul pourrait ne pas être suffisant.
Les cancers aéro-digestifs acquièrent généralement
des anomalies dans plus d'une voie de signalisation au cours de la
carcinogenèse, et l'EGFR seul n'est qu'une des nombreuses cibles dans
un système complexe. Le ciblage de ce récepteur pourrait donc
plutôt devenir la composante d'une stratégie double ou
multicible, destinée à traiter la complexité des voies de
signalisation et à empêcher l'émergence de
résistance. Les réseaux d'interaction de l'EGFR avec la voie des
récepteurs VEGF/VEGF sont bien établis. La résistance
préclinique et clinique aux inhibiteurs d'EGFR a été
attribuée à l'augmentation des taux de VEGF.134 En
conséquence, l'association d'inhibiteurs d'EGFR et d'agents
anti-angiogenèse apparaît comme une nouvelle stratégie
thérapeutique séduisante. Des doubles inhibiteurs des
récepteurs EGFR et VEGF, comme le vandetanib, ont également
été
développés.135
Le profilage moléculaire des cancers des VADS peut
considérablement améliorer les résultats des traitements
anti-EGFR, en permettant d'identifier les tumeurs sensibles à cette
stratégie. La réalisation d'études cliniques dans des
populations de patients développant certaines caractéristiques
moléculaires est plus susceptible de produire les résultats
souhaités. En outre, le prélèvement de tissu tumoral
pré et post-traitement pourrait permettre de mieux comprendre les
mécanismes d'action et de résistance aux nouvelles
substances.
L'effet biologique des inhibiteurs d'EGFR a également
été évalué dans des prélèvements
biopsiques de la peau, qui constitue un tissu de substitution facilement
accessible; cependant, les résultats des biopsies cutanées et
tumorales peuvent être divergents.136 La validation de critères
pharmacodynamiques appropriés, qui pourraient guider la posologie et
l'évaluation de la réponse, reste d'une suprême importance
dans le développement des stratégies anti-EGFR. En outre, du
fait que la stabilisation tumorale -pas seulement la régression- est
liée à l'efficacité anti-tumorale des inhibiteurs d'EGFR,
il serait souhaitable d'inclure le délai à la progression dans
les critères d'évaluation des premières phases
d'études cliniques. Enfin, il pourrait être important
d'étudier la séquence de traitement la plus appropriée
pour les inhibiteurs d'EGFR et la chimiothérapie. Il a
été suggéré que de meilleurs résultats
pourraient être obtenus par une administration intermittente des agents
ciblant l'EGFR lorsqu'ils sont associés à des produits
chimiothérapiques.137
Des études cliniques sur les TKI-EGFR dans le CBNPC avancé sont
en cours pour tester ces hypothèses.
Les traitements anti-EGFR deviennent une partie intégrante de
l'arsenal thérapeutique anticancéreux et sont actuellement
évalués dans divers contextes cliniques, notamment en
association avec la chimiothérapie et la radiothérapie dans les
maladies potentiellement curables. Dans le même temps, de nouveaux
inhibiteurs de l'EGFR et des stratégies multicibles sont en cours
d'évaluation dans les cancers des VADS et d'autres tumeurs malignes.
Une meilleure compréhension de la biologie des cancers des VADS devrait
favoriser l'optimisation des stratégies anti-EGFR et des autres
thérapies ciblées.
Informations sur les auteurs
Correspondance: Athanassios Argiris, MD, Division of
Hematology-Oncology, University of Pittsburgh, UPMC Cancer Pavilion, Fifth
Floor, Pittsburgh, PA 15232
(argirisae{at}upmc.edu).
FMC disponible en ligne à
www.jama.com
Rédacteur en chef de la Revue Clinique: Michael S. Lauer, MD.
Nous encourageons les auteurs à soumettre leurs articles en vue de
publication dans la section Revue Clinique. Vous pouvez contacter Michael S.
Lauer, MD, à
michael.lauer{at}jama-archives.org.
Contributions des auteurs: Le Dr Karamouzis a eu un accès
complet à toutes les données de l'étude et accepte la
responsabilité de l'intégrité des données et de la
précision de l'analyse des données.
Conception et schéma de l'étude: Karamouzis, Grandis,
Argiris.
Recueil des données: Karamouzis, Grandis, Argiris.
Analyse et interprétation des données: Karamouzis,
Argiris.
Rédaction du manuscrit: Karamouzis, Argiris.
Revue critique du manuscrit: Karamouzis, Grandis, Argiris.
Aide administrative, technique et matérielle: Karamouzis, Grandis,
Argiris.
Liens financiers: Le Dr Grandis a déclaré avoir
reçu des honoraires de Bristol-Myers Squibb et Astra-Zeneca et
bénéficié d'un soutien de recherche du National Cancer
Institute et de OSI Pharmaceuticals. Le Dr Argiris a déclaré
avoir travaillé comme consultant pour Amgen; avoir reçu un
soutien pour ses recherches du National Cancer Institute et d'Astra-Zeneca,
Bristol-Myers Squibb, Genentech, Lilly Oncology, Millennium Pharmaceuticals,
M's Science Corporation, Novartis, et Pfizer; et d'avoir reçu des
honoraires d' Astra-Zeneca et Genentech. Aucun autre lien financier n'a
été déclaré.
Financement/Soutien: Ce travail a bénéficié du
soutien partiel du National Cancer Institute, Head and Neck SPORE: P50
CA097190-01A1, et Lung SPORE: P50 CA90440-06.
Rôle des sponsors: L'organisation de financement n'a
joué aucun rôle dans le schéma, la conduite, le recueil,
l'analyse, l'interprétation des données, ni dans la
préparation, la revue ou l'approbation du manuscrit.
Affiliations des auteurs: Division of Hematology-Oncology,
Department of Medicine, Department of Otolaryngology, and Head and Neck Cancer
Program of the University of Pittsburgh Cancer Institute, University of
Pittsburgh, Pittsburgh, Pennsylvania.
BIBLIOGRAPHIE
| |
1. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence,
mortality, and prevalence across five continents: defining priorities to
reduce cancer disparities in different geographic regions of the world.
J Clin Oncol.2006; 24(14):2137
-2150.
FREE FULL TEXT
2. Hanahan D, Weinberg RA. The hallmarks of cancer.
Cell.2000; 100(1):57
-70.
PUBMED
3. Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting
in cancer. Semin Oncol.2006; 33(4):369
-385.
PUBMED
4. Fontanini G, Vignati S, Bigini D, et al. Epidermal growth factor
receptor (EGFr) expression in non-small cell lung carcinomas correlates with
metastatic involvement of hilar and mediastinal lymph nodes in the squamous
subtype. Eur J Cancer.1995; 31A(2):178
-183.
5. Hirsch FR, Varella-Garcia M, Bunn PA Jr, et al. Epidermal growth
factor receptor in non-small-cell lung carcinomas: correlation between gene
copy number and protein expression and impact on prognosis. J Clin
Oncol.2003; 21(20):3798
-3807.
FREE FULL TEXT
6. Rusch V, Baselga J, Cordon-Cardo C, et al. Differential expression
of the epidermal growth factor receptor and its ligands in primary non-small
cell lung cancers and adjacent benign lung. Cancer
Res.1993; 53(10)(suppl):2379
-2385.
FREE FULL TEXT
7. Brabender J, Danenberg KD, Metzger R, et al. Epidermal growth
factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is
correlated with survival. Clin Cancer Res.2001; 7(7):1850
-1855.
FREE FULL TEXT
8. Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor
receptor expression on survival and pattern of relapse in patients with
advanced head and neck carcinoma. Cancer Res.2002; 62(24):7350
-7356.
FREE FULL TEXT
9. Hanawa M, Suzuki S, Dobashi Y, et al. EGFR protein overexpression
and gene amplification in squamous cell carcinomas of the esophagus.
Int J Cancer.2006; 118(5):1173
-1180.
PUBMED
10. Gibault L, Metges JP, Conan-Charlet V, et al. Diffuse EGFR staining
is associated with reduced overall survival in locally advanced oesophageal
squamous cell cancer. Br J Cancer.2005; 93(1):107
-115.
PUBMED
11. Wang KL, Wu TT, Choi IS, et al. Expression of epidermal growth
factor receptor in esophageal and esophagogastric junction adenocarcinomas:
association with poor outcome. Cancer.2007; 109(4):658
-667.
PUBMED
12. Scagliotti GV, Selvaggi G, Novello S, Hirsch FR. The biology of
epidermal growth factor receptor in lung cancer. Clin Cancer
Res.2004; 10(12, pt 2):4227s
-4232s.
13. Meert AP, Martin B, Delmotte P, et al. The role of EGF-R expression
on patient survival in lung cancer: a systematic review with meta-analysis.
Eur Respir J.2002; 20(4):975
-981.
FREE FULL TEXT
14. Rusch V, Klimstra D, Venkatraman E, Pisters PW, Langenfeld J,
Dmitrovsky E. Overexpression of the epidermal growth factor receptor and its
ligand transforming growth factor alpha is frequent in resectable non-small
cell lung cancer but does not predict tumor progression. Clin
Cancer Res.1997; 3(4):515
-522.
ABSTRACT
15. Fujino S, Enokibori T, Tezuka N, et al. A comparison of epidermal
growth factor receptor levels and other prognostic parameters in non-small
cell lung cancer. Eur J Cancer.1996; 32A(12):2070
-2074.
16. Pfeiffer P, Clausen PP, Andersen K, Rose C. Lack of prognostic
significance of epidermal growth factor receptor and the oncoprotein p185HER-2
in patients with systemically untreated non-small-cell lung cancer: an
immunohistochemical study on cryosections. Br J
Cancer.1996; 74(1):86
-91.
PUBMED
17. Pastorino U, Andreola S, Tagliabue E, et al. Immunocytochemical
markers in stage I lung cancer: relevance to prognosis. J Clin
Oncol.1997; 15(8):2858
-2865.
ABSTRACT
18. Volm M, Rittgen W, Drings P. Prognostic value of ERBB-1, VEGF,
cyclin A, FOS, JUN and MYC in patients with squamous cell lung carcinomas.
Br J Cancer.1998; 77(4):663
-669.
PUBMED
19. Rubin Grandis J, Melhem MF, Gooding WE, et al. Levels of TGF-alpha
and EGFR protein in head and neck squamous cell carcinoma and patient
survival. J Natl Cancer Inst.1998; 90(11):824
-832.
FREE FULL TEXT
20. Ozawa S, Ueda M, Ando N, Shimizu N, Abe O. Prognostic significance
of epidermal growth factor receptor in esophageal squamous cell carcinomas.
Cancer.1989; 63(11):2169
-2173.
PUBMED
21. Chung CH, Ely K, McGavran L, et al. Increased epidermal growth
factor receptor gene copy number is associated with poor prognosis in head and
neck squamous cell carcinomas. J Clin Oncol.2006; 24(25):4170
-4176.
FREE FULL TEXT
22. Marshall J. Clinical implications of the mechanism of epidermal
growth factor receptor inhibitors. Cancer.2006; 107(6):1207
-1218.
PUBMED
23. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor
biology in head and neck cancer. J Clin Oncol.2006; 24(17):2666
-2672.
FREE FULL TEXT
24. Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level.
Nat Rev Mol Cell Biol.2006; 7(7):505
-516.
PUBMED
25. Adjei AA. Novel combinations based on epidermal growth factor
receptor inhibition. Clin Cancer Res.2006; 12(14, pt 2):4446s
-4450s.
FREE FULL TEXT
26. Toker A, Yoeli-Lerner M. Akt signaling and cancer: surviving but
not moving on. Cancer Res.2006; 66(8):3963
-3966.
FREE FULL TEXT
27. Leeman RJ, Lui VW, Grandis JR. STAT3 as a therapeutic target in
head and neck cancer. Expert Opin Biol Ther.2006; 6(3):231
-241.
PUBMED
28. Thomas SM, Coppelli FM, Wells A, et al. Epidermal growth factor
receptor-stimulated activation of phospholipase Cgamma-1 promotes invasion of
head and neck squamous cell carcinoma. Cancer Res.2003; 63(17):5629
-5635.
FREE FULL TEXT
29. Thomas SM, Bhola NE, Zhang Q, et al. Cross-talk between G
protein-coupled receptor and epidermal growth factor receptor signaling
pathways contributes to growth and invasion of head and neck squamous cell
carcinoma. Cancer Res.2006; 66(24):11831
-11839.
FREE FULL TEXT
30. Ingram JL, Bonner JC. EGF and PDGF receptor tyrosine kinases as
therapeutic targets for chronic lung diseases. Curr Mol
Med.2006; 6(4):409
-421.
PUBMED
31. Karamouzis MV, Papavassiliou AG. The IGF-1 network in lung
carcinoma therapeutics. Trends Mol Med.2006; 12(12):595
-602.
PUBMED
32. Gururaj AE, Rayala SK, Vadlamudi RK, Kumar R. Novel mechanisms of
resistance to endocrine therapy: genomic and nongenomic considerations.
Clin Cancer Res.2006; 12(3, pt 2):1001s
-1007s.
FREE FULL TEXT
33. Frey MR, Dise RS, Edelblum KL, Polk DB. p38 kinase regulates
epidermal growth factor receptor down-regulation and cellular migration.
EMBO J.2006; 25(24):5683
-5692.
PUBMED
34. Wiley HS. Trafficking of the ErbB receptors and its influence on
signaling. Exp Cell Res.2003; 284(1):78
-88.
PUBMED
35. Oliveira S, van Bergen en Henegouwen PM, Storm G, Schiffelers RM.
Molecular biology of epidermal growth factor receptor inhibition for cancer
therapy. Expert Opin Biol Ther.2006; 6(6):605
-617.
PUBMED
36. Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth
factor receptor (EGFRvIII) contributes to head and neck cancer growth and
resistance to EGFR targeting. Clin Cancer Res.2006; 12(17):5064
-5073.
FREE FULL TEXT
37. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the
epidermal growth factor receptor underlying responsiveness of non-small-cell
lung cancer to gefitinib. N Engl J Med.2004; 350(21):2129
-2139.
PUBMED
38. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer:
correlation with clinical response to gefitinib therapy.
Science.2004; 304(5676):1497
-1500.
FREE FULL TEXT
39. Guo M, Liu S, Lu F. Gefitinib-sensitizing mutations in esophageal
carcinoma. N Engl J Med.2006; 354(20):2193
-2194.
PUBMED
40. Willmore-Payne C, Holden JA, Layfield LJ. Detection of EGFR- and
HER2-activating mutations in squamous cell carcinoma involving the head and
neck. Mod Pathol.2006; 19(5):634
-640.
PUBMED
41. Baselga J. Targeting tyrosine kinases in cancer: the second wave.
Science.2006; 312(5777):1175
-1178.
FREE FULL TEXT
42. Lacouture ME. Mechanisms of cutaneous toxicities to EGFR
inhibitors. Nat Rev Cancer.2006; 6(10):803
-812.
PUBMED
43. Tejpar S, Piessevaux H, Claes K, et al. Magnesium wasting
associated with epidermal-growth-factor receptor-targeting antibodies in
colorectal cancer: a prospective study. Lancet Oncol.2007; 8(5):387
-394.
PUBMED
44. Schrag D, Chung KY, Flombaum C, Saltz L. Cetuximab therapy and
symptomatic hypomagnesemia. J Natl Cancer Inst.2005; 97(16):1221
-1224.
FREE FULL TEXT
45. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM.
Structural basis for inhibition of the epidermal growth factor receptor by
cetuximab. Cancer Cell.2005; 7(4):301
-311.
PUBMED
46. Imai K, Takaoka A. Comparing antibody and small-molecule therapies
for cancer. Nat Rev Cancer.2006; 6(9):714
-727.
PUBMED
47. Kawaguchi Y, Kono K, Mimura K, Sugai H, Akaike H, Fujii H.
Cetuximab induce antibody-dependent cellular cytotoxicity against
EGFR-expressing esophageal squamous cell carcinoma. Int J
Cancer.2007; 120(4):781
-787.
PUBMED
48. Govindan SV, Griffiths GL, Hansen HJ, Horak ID, Goldenberg DM.
Cancer therapy with radiolabeled and drug/toxin-conjugated antibodies.
Technol Cancer Res Treat.2005; 4(4):375
-391.
PUBMED
49. Heymach JV, Nilsson M, Blumenschein G, Papadimitrakopoulou V,
Herbst R. Epidermal growth factor receptor inhibitors in development for the
treatment of non-small cell lung cancer. Clin Cancer
Res.2006; 12(14, pt 2):4441s
-4445s.
FREE FULL TEXT
50. Carter TA, Wodicka LM, Shah NP, et al. Inhibition of drug-resistant
mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U
S A.2005; 102(31):11011
-11016.
FREE FULL TEXT
51. Verreault M, Webb MS, Ramsay EC, Bally MB. Gene silencing in the
development of personalized cancer treatment: the targets, the agents and the
delivery systems. Curr Gene Ther.2006; 6(4):505
-533.
PUBMED
52. Coppelli FM, Grandis JR. Oligonucleotides as anticancer agents:
from the benchside to the clinic and beyond. Curr Pharm
Des.2005; 11(22):2825
-2840.
PUBMED
53. Giaccone G. Epidermal growth factor receptor inhibitors in the
treatment of non-small-cell lung cancer. J Clin Oncol.2005; 23(14):3235
-3242.
FREE FULL TEXT
54. Patel JD, Pasche B, Argiris A. Targeting non-small cell lung cancer
with epidermal growth factor tyrosine kinase inhibitors: where do we stand,
where do we go. Crit Rev Oncol Hematol.2004; 50(3):175
-186.
PUBMED
55. Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional
randomized phase II trial of gefitinib for previously treated patients with
advanced non-small-cell lung cancer (The IDEAL 1 Trial) [published correction
appears in J Clin Oncol. 2004;22(23):4811]. J Clin
Oncol.2003; 21(12):2237
-2246.
FREE FULL TEXT
56. Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an
inhibitor of the epidermal growth factor receptor tyrosine kinase, in
symptomatic patients with non-small cell lung cancer: a randomized trial.
JAMA.2003; 290(16):2149
-2158.
FREE FULL TEXT
57. Pérez-Soler R, Chachoua A, Hammond LA, et al. Determinants
of tumor response and survival with erlotinib in patients with non-small-cell
lung cancer. J Clin Oncol.2004; 22(16):3238
-3247.
FREE FULL TEXT
58. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in
previously treated non-small-cell lung cancer. N Engl J
Med.2005; 353(2):123
-132.
PUBMED
59. Herbst RS, Prager D, Hermann R, et al. TRIBUTE: a phase III trial
of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel
chemotherapy in advanced non-small-cell lung cancer. J Clin
Oncol.2005; 23(25):5892
-5899.
FREE FULL TEXT
60. Gatzemeier U, Pluzanska A, Szczesna A, et al. Phase III study of
erlotinib in combination with cisplatin and gemcitabine in advanced
non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial.
J Clin Oncol.2007; 25(12):1545
-1552.
FREE FULL TEXT
61. Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best
supportive care in previously treated patients with refractory advanced
non-small-cell lung cancer: results from a randomised, placebo-controlled,
multi-centre study (Iressa Survival Evaluation in Lung Cancer).
Lancet.2005; 366(9496):1527
-1537.
PUBMED
62. Herbst RS, Giaccone G, Schiller JH, et al. Gefitinib in combination
with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a
phase III trial—INTACT 2. J Clin Oncol.2004; 22(5):785
-794.
FREE FULL TEXT
63. Giaccone G, Herbst RS, Manegold C, et al. Gefitinib in combination
with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase
III trial—INTACT 1. J Clin Oncol.2004; 22(5):777
-784.
FREE FULL TEXT
64. Kelly K, Gaspar L, Chansky K, Albain K, Crowley J, Gandara D. Low
incidence of pneumonitis on SWOG 0023: a preliminary analysis of an ongoing
phase III trial of concurrent chemoradiotherapy followed by consolidation
docetaxel and Iressa/placebo maintenance in patients with inoperable stage III
non-small cell lung cancer. J Clin Oncol.2005; 23(16)(suppl):7058
.
65. Stewart S, Cohen E, Licitra L, et al. A phase III randomized
parallel-group study of gefitinib (Iressa) versus methotrexate (IMEX) in
patients with recurrent squamous cell carcinoma of the head and neck.
Proc Am Assoc Cancer Res. 2007:A3522
.66. Burtness B, Goldwasser MA, Flood W, Mattar B, Forastiere AA. Phase
III randomized trial of cisplatin plus placebo compared with cisplatin plus
cetuximab in metastatic/recurrent head and neck cancer: an Eastern Cooperative
Oncology Group study. J Clin Oncol.2005; 23(34):8646
-8654.
FREE FULL TEXT
67. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab
for squamous-cell carcinoma of the head and neck. N Engl J
Med.2006; 354(6):567
-578.
PUBMED
68. Bezjak A, Tu D, Seymour L, et al. Symptom improvement in lung
cancer patients treated with erlotinib: quality of life analysis of the
National Cancer Institute of Canada Clinical Trials Group Study BR.21.
J Clin Oncol.2006; 24(24):3831
-3837.
FREE FULL TEXT
69. Akita RW, Sliwkowski MX. Preclinical studies with erlotinib
(Tarceva). Semin Oncol.2003; 30(3)(suppl 7):15
-24.
PUBMED
70. Pedersen MW, Pedersen N, Ottesen LH, Poulsen HS. Differential
response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII.
Br J Cancer.2005; 93(8):915
-923.
PUBMED
71. Gandara DR, Gumerlock PH. Epidermal growth factor receptor tyrosine
kinase inhibitors plus chemotherapy: case closed or is the jury still out?
J Clin Oncol.2005; 23(25):5856
-5858.
FREE FULL TEXT
72. Hanna N, Lilenbaum R, Ansari R, et al. Phase II trial of cetuximab
in patients with previously treated non-small-cell lung cancer. J
Clin Oncol.2006; 24(33):5253
-5258.
FREE FULL TEXT
73. Grandis JR, Tweardy DJ. Elevated levels of transforming growth
factor alpha and epidermal growth factor receptor messenger RNA are early
markers of carcinogenesis in head and neck cancer. Cancer
Res.1993; 53(15):3579
-3584.
FREE FULL TEXT
74. Cohen EE, Kane MA, List MA, et al. Phase II trial of gefitinib 250
mg daily in patients with recurrent and/or metastatic squamous cell carcinoma
of the head and neck. Clin Cancer Res.2005; 11(23):8418
-8424.
FREE FULL TEXT
75. Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in
recurrent or metastatic squamous cell carcinoma of the head and neck.
J Clin Oncol.2003; 21(10):1980
-1987.
FREE FULL TEXT
76. Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL.
Multicenter phase II study of erlotinib, an oral epidermal growth factor
receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic
squamous cell cancer of the head and neck. J Clin
Oncol.2004; 22(1):77
-85.
FREE FULL TEXT
77. Kirby AM, A'Hern RP, D'Ambrosio C, et al. Gefitinib (ZD1839,
Iressa) as palliative treatment in recurrent or metastatic head and neck
cancer. Br J Cancer.2006; 94(5):631
-636.
PUBMED
78. Abidoye OO, Cohen EE, Wong SJ, et al. A phase II study of lapatinib
(GW572016) in recurrent/metastatic (R/M) squamous cell carcinoma of the head
and neck (SCCHN). J Clin Oncol.2006; 24(18)(suppl):5568
.
79. Cohen EE. Role of epidermal growth factor receptor pathway-targeted
therapy in patients with recurrent and/or metastatic squamous cell carcinoma
of the head and neck. J Clin Oncol.2006; 24(17):2659
-2665.
FREE FULL TEXT
80. Pfister DG, Su YB, Kraus DH, et al. Concurrent cetuximab,
cisplatin, and concomitant boost radiotherapy for locoregionally advanced,
squamous cell head and neck cancer: a pilot phase II study of a new
combined-modality paradigm. J Clin Oncol.2006; 24(7):1072
-1078.
FREE FULL TEXT
81. Trigo J, Hitt R, Koralewski P, et al. Cetuximab monotherapy is
active in patients (pts) with platinum-refractory recurrent/metastatic
squamous cell carcinoma of the head and neck (SCCHN): results of a phase II
study. J Clin Oncol.2004; 22(14)(suppl):5502
.
82. Baselga J, Trigo JM, Bourhis J, et al. Phase II multicenter study
of the antiepidermal growth factor receptor monoclonal antibody cetuximab in
combination with platinum-based chemotherapy in patients with
platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the
head and neck. J Clin Oncol.2005; 23(24):5568
-5577.
FREE FULL TEXT
83. Herbst RS, Arquette M, Shin DM, et al. Phase II multicenter study
of the epidermal growth factor receptor antibody cetuximab and cisplatin for
recurrent and refractory squamous cell carcinoma of the head and neck.
J Clin Oncol.2005; 23(24):5578
-5587.
FREE FULL TEXT
84. Vermorken J, Mesia R, Vega-Villegas M, et al. Cetuximab in
combination with cisplatin or carboplatin and 5-fluorouracil (5-FU) in the
first-line treatment of patients with recurrent and/or metastatic squamous
cell carcinoma of the head and neck (R&M SCCHN) (EXTREME). J
Clin Oncol.2006; 24(18)(suppl):5537
.
85. Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal
cancer. Semin Radiat Oncol.2007; 17(1):2
-9.
PUBMED
86. Janmaat ML, Gallegos-Ruiz MI, Rodriguez JA, et al. Predictive
factors for outcome in a phase II study of gefitinib in second-line treatment
of advanced esophageal cancer patients. J Clin Oncol.2006; 24(10):1612
-1619.
FREE FULL TEXT
87. Sudo T, Mimori K, Nagahara H, et al. Identification of EGFR
mutations in esophageal cancer. Eur J Surg Oncol.2007; 33(1):44
-48.
PUBMED
88. Kwak EL, Jankowski J, Thayer SP, et al. Epidermal growth factor
receptor kinase domain mutations in esophageal and pancreatic adenocarcinomas.
Clin Cancer Res.2006; 12(14, pt 1):4283
-4287.
FREE FULL TEXT
89. Dragovich T, McCoy S, Fenoglio-Preiser CM, et al. Phase II trial of
erlotinib in gastroesophageal junction and gastric adenocarcinomas: SWOG 0127.
J Clin Oncol.2006; 24(30):4922
-4927.
FREE FULL TEXT
90. Enzinger PC, Yock T, Suh W, et al. Phase II cisplatin, irinotecan,
cetuximab and concurrent radiation therapy followed by surgery for locally
advanced esophageal cancer. J Clin Oncol.2006; 24(18) (suppl):4064
.
91. Dobelbower MC, Russo SM, Raisch KP, et al. Epidermal growth factor
receptor tyrosine kinase inhibitor, erlotinib, and concurrent 5-fluorouracil,
cisplatin and radiotherapy for patients with esophageal cancer: a phase I
study. Anticancer Drugs.2006; 17(1):95
-102.
PUBMED
92. Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the
epidermal growth factor receptor and in KRAS are predictive and prognostic
indicators in patients with non-small-cell lung cancer treated with
chemotherapy alone and in combination with erlotinib. J Clin
Oncol.2005; 23(25):5900
-5909.
FREE FULL TEXT
93. Pham D, Kris MG, Riely GJ, et al. Use of cigarette-smoking history
to estimate the likelihood of mutations in epidermal growth factor receptor
gene exons 19 and 21 in lung adenocarcinomas. J Clin
Oncol.2006; 24(11):1700
-1704.
FREE FULL TEXT
94. West HL, Franklin WA, McCoy J, et al. Gefitinib therapy in advanced
bronchioloalveolar carcinoma: Southwest Oncology Group Study S0126.
J Clin Oncol.2006; 24(12):1807
-1813.
FREE FULL TEXT
95. Raz DJ, He B, Rosell R, Jablons DM. Current concepts in
bronchioloalveolar carcinoma biology. Clin Cancer Res.2006; 12(12):3698
-3704.
FREE FULL TEXT
96. Pérez-Soler R, Saltz L. Cutaneous adverse effects with
HER1/EGFR-targeted agents: is there a silver lining? J Clin
Oncol.2005; 23(22):5235
-5246.
FREE FULL TEXT
97. Amador ML, Oppenheimer D, Perea S, et al. An epidermal growth
factor receptor intron 1 polymorphism mediates response to epidermal growth
factor receptor inhibitors. Cancer Res.2004; 64(24):9139
-9143.
FREE FULL TEXT
98. Calvo E, Baselga J. Ethnic differences in response to epidermal
growth factor receptor tyrosine kinase inhibitors. J Clin
Oncol.2006; 24(14):2158
-2163.
FREE FULL TEXT
99. Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth
factor receptor signaling pathway in lung cancers. Int J
Cancer.2006; 118(2):257
-262.
PUBMED
100. Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, et al. Low
incidence of mutations in EGFR kinase domain in Caucasian patients with head
and neck squamous cell carcinoma. Eur J Cancer.2006; 42(1):109
-111.
PUBMED
101. Mitsudomi T, Kosaka T, Endoh H, et al. Mutations of the epidermal
growth factor receptor gene predict prolonged survival after gefitinib
treatment in patients with non-small-cell lung cancer with postoperative
recurrence. J Clin Oncol.2005; 23(11):2513
-2520.
FREE FULL TEXT
102. Han SW, Kim TY, Hwang PG, et al. Predictive and prognostic impact
of epidermal growth factor receptor mutation in non-small-cell lung cancer
patients treated with gefitinib. J Clin Oncol.2005; 23 (11):2493
-2501.
FREE FULL TEXT
103. Taron M, Ichinose Y, Rosell R, et al. Activating mutations in the
tyrosine kinase domain of the epidermal growth factor receptor are associated
with improved survival in gefitinib-treated chemorefractory lung
adenocarcinomas. Clin Cancer Res.2005; 11 (16):5878
-5885.
FREE FULL TEXT
104. Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung
cancer—molecular and clinical predictors of outcome. N Engl J
Med.2005; 353(2):133
-144.
PUBMED
105. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and
resistance of non-small-cell lung cancer to gefitinib. N Engl J
Med.2005; 352(8):786
-792.
PUBMED
106. Wirth LJ, Haddad RI, Lindeman NI, et al. Phase I study of gefitinib
plus celecoxib in recurrent or metastatic squamous cell carcinoma of the head
and neck. J Clin Oncol.2005; 23(28):6976
-6981.
FREE FULL TEXT
107. Inoue A, Suzuki T, Fukuhara T, et al. Prospective phase II study of
gefitinib for chemotherapynaive patients with advanced non-small-cell lung
cancer with epidermal growth factor receptor gene mutations. J Clin
Oncol.2006; 24(21):3340
-3346.
FREE FULL TEXT
108. Asahina H, Yamazaki K, Kinoshita I, et al. A phase II trial of
gefitinib as first-line therapy for advanced non-small cell lung cancer with
epidermal growth factor receptor mutations. Br J
Cancer.2006; 95(8):998
-1004.
PUBMED
109. Paz-Ares L, Sanchez JM, Garcia-Velasco A, et al. A prospective
phase II trial of erlotinib in advanced non-small cell lung cancer (NSCLC)
patients with mutations in the tyrosine kinase (TK) domain of the epidermal
growth factor receptor (EGFR). J Clin Oncol.2006; 24(18)(suppl):7020
.
110. Kimura H, Fujiwara Y, Sone T, et al. High sensitivity detection of
epidermal growth factor receptor mutations in the pleural effusion of
non-small cell lung cancer patients. Cancer Sci.2006; 97(7):642
-648.
PUBMED
111. Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor
receptor gene and protein and gefitinib sensitivity in non-small-cell lung
cancer. J Natl Cancer Inst.2005; 97(9):643
-655.
FREE FULL TEXT
112. Hirsch FR, Varella-Garcia M, McCoy J, et al. Increased epidermal
growth factor receptor gene copy number detected by fluorescence in situ
hybridization associates with increased sensitivity to gefitinib in patients
with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study.
J Clin Oncol.2005; 23(28):6838
-6845.
FREE FULL TEXT
113. Hirsch FR, Varella-Garcia M, Bunn PA Jr, et al. Molecular
predictors of outcome with gefitinib in a phase III placebo-controlled study
in advanced non-small-cell lung cancer. J Clin Oncol.2006; 24(31):5034
-5042.
FREE FULL TEXT
114. Cappuzzo F, Toschi L, Domenichini I, et al. HER3 genomic gain and
sensitivity to gefitinib in advanced non-small-cell lung cancer patients.
Br J Cancer.2005; 93(12):1334
-1340.
PUBMED
115. Cappuzzo F, Varella-Garcia M, Shigematsu H, et al. Increased HER2
gene copy number is associated with response to gefitinib therapy in epidermal
growth factor receptor-positive non-small-cell lung cancer patients.
J Clin Oncol.2005; 23(22):5007
-5018.
FREE FULL TEXT
116. Ichihara S, Toyooka S, Fujiwara Y, et al. The impact of epidermal
growth factor receptor gene status on gefitinib-treated Japanese patients with
non-small-cell lung cancer. Int J Cancer.2007; 120(6):1239
-1247.
PUBMED
117. Liu W, Innocenti F, Chen P, Das S, Cook EH Jr, Ratain MJ.
Interethnic difference in the allelic distribution of human epidermal growth
factor receptor intron 1 polymorphism. Clin Cancer
Res.2003; 9(3):1009
-1012.
FREE FULL TEXT
118. Araújo A, Ribeiro R, Azevedo I, et al. Genetic polymorphisms
of the epidermal growth factor and related receptor in non-small cell lung
cancer—a review of the literature. Oncologist.2007; 12(2):201
-210.
FREE FULL TEXT
119. Li J, Karlsson MO, Brahmer J, et al. CYP3A phenotyping approach to
predict systemic exposure to EGFR tyrosine kinase inhibitors. J
Natl Cancer Inst.2006; 98(23):1714
-1723.
FREE FULL TEXT
120. Dziadziuszko R, Hirsch FR, Varella-Garcia M, Bunn PA Jr. Selecting
lung cancer patients for treatment with epidermal growth factor receptor
tyrosine kinase inhibitors by immunohistochemistry and fluorescence in situ
hybridization—why, when, and how? Clin Cancer
Res.2006; 12(14, pt 2):4409s
-4415s.
FREE FULL TEXT
121. Bailey LR, Kris M, Wolf M, et al. Tumor epidermal growth factor
receptor (EGFR) expression levels does not predict for response in patients
(pts) receiving gefitinib ("Iressa," ZD1839) monotherapy for
pretreated advanced non-small-cell lung cancer (NSCLC): IDEAL 1 and 2.
Proc Am Assoc Cancer Res. 2003:A1362
.122. Toschi L, Cappuzzo F. Understanding the new genetics of
responsiveness to epidermal growth factor receptor tyrosine kinase inhibitors.
Oncologist.2007; 12(2):211
-220.
FREE FULL TEXT
123. Cappuzzo F, Toschi L, Tallini G, et al. Insulin-like growth factor
receptor 1 (IGFR-1) is significantly associated with longer survival in
non-small-cell lung cancer patients treated with gefitinib. Ann
Oncol.2006; 17(7):1120
-1127.
FREE FULL TEXT
124. Cappuzzo F, Magrini E, Ceresoli GL, et al. Akt phosphorylation and
gefitinib efficacy in patients with advanced non-small-cell lung cancer.
J Natl Cancer Inst.2004; 96(15):1133
-1141.
FREE FULL TEXT
125. Noro R, Gemma A, Kosaihira S, et al. Gefitinib (IRESSA) sensitive
lung cancer cell lines show phosphorylation of Akt without ligand stimulation.
BMC Cancer.2006; 6(1):277
.
PUBMED
126. Sergina NV, Rausch M, Wang D, et al. Escape from HER-family
tyrosine kinase inhibitor therapy by the kinase-inactive HER3.
Nature. 2007;445
(7126): 437-441.
PUBMED
127. Toyooka S, Tokumo M, Shigematsu H, et al. Mutational and epigenetic
evidence for independent path-ways for lung adenocarcinomas arising in smokers
and never smokers. Cancer Res.2006; 66(3):1371
-1375.
FREE FULL TEXT
128. Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao MS. K-ras mutations
in non-small-cell lung carcinoma: a review. Clin Lung
Cancer.2006; 8(1):30
-38.
PUBMED
129. Pao W, Miller VA. Epidermal growth factor receptor mutations,
small-molecule kinase inhibitors, and non-small-cell lung cancer: current
knowledge and future directions. J Clin Oncol.2005; 23(11):2556
-2568.
FREE FULL TEXT
130. Miller VA, Zakowski M, Riely GJ, et al. EGFR mutations and copy
number, EGFR expression and KRAS mutation as predictors of outcome with
erlotinib in bronchioalveolar cell carcinoma (BAC): results of a prospective
study. J Clin Oncol.2006; 24(18)(suppl):364
.
131. Tsao M, Zhu C, Sakurada A, et al. An analysis of the prognostic and
predictive importance of K-ras mutation status in the National Cancer
Institute of Canada Clinical Trials Group BR.21 study of erlotinib versus
placebo in the treatment of non-small cell lung cancer. J Clin
Oncol.2006; 24(18)(suppl):365
.
132. Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM.
Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR):
combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer
Res.2004; 64(15):5355
-5362.
FREE FULL TEXT
133. Baselga J, Schöffski P, Rojo F, et al. A phase I
pharmacokinetic (PK) and molecular pharmacodynamic (PD) study of the
combination of two anti-EGFR therapies, the monoclonal antibody (MAb)
cetuximab (C) and the tyrosine kinase inhibitor (TKI) gefitinib (G), in
patients (pts) with advanced colorectal (CRC), head and neck (HNC) and
non-small cell lung cancer (NSCLC). J Clin Oncol.2006; 24(18) (suppl):3006
.
134. Viloria-Petit A, Crombet T, Jothy S, et al. Acquired resistance to
the antitumor effect of epidermal growth factor receptor-blocking antibodies
in vivo: a role for altered tumor angiogenesis. Cancer
Res.2001; 61(13):5090
-5101.
FREE FULL TEXT
135. Bozec A, Fischel JL, Milano G. Epidermal growth factor
receptor/angiogenesis dual targeting: preclinical experience. Curr
Opin Oncol.2006; 18(4):330
-334.
PUBMED
136. Calvo E, Malik S, Siu L, et al. Assessment of erlotinib
pharmacodynamics in tumors and skin of patients with head and neck cancer.
Ann Oncol.2007; 18(4):761
-767.
FREE FULL TEXT
137. Solit DB, She Y, Lobo J, et al. Pulsatile administration of the
epidermal growth factor receptor inhibitor gefitinib is significantly more
effective than continuous dosing for sensitizing tumors to paclitaxel.
Clin Cancer Res.2005; 11(5):1983
-1989.
FREE FULL TEXT
ARTICLE EN RAPPORT
Cette semaine dans le JAMA
JAMA. 2007;298:9.
Texte Complet
|