Anesthetic Activation of Nociceptors: Adding Insult to Injury?
General anesthetics induce loss of consciousness at low concentrations and produce a lack of response to painful stimuli at high concentrations (1). For inhaled anesthetics, this endpoint has traditionally been observed as the lack of withdrawal to skin incision and has been used to compare their anesthetic potency (2). It is unclear, however, whether many general anesthetics possess analgesic properties in the sense of acting specifically on targets in the pain pathway. In fact, the contrary may be true, because low concentrations of inhaled anesthetics reportedly cause hyperalgesia (3–6).
At first, these reports seem surprising and counterintuitive. One might expect that no pain is felt during surgery under general anesthesia, which leads to the assumption that general anesthetics must have analgesic properties. Moreover, the notion that anesthesia results from a decrease of neuronal excitability seems at odds with their excitatory effects. Nevertheless, clinical use of general anesthetics has indicated their ability to increase excitability in several systems. For example, some intravenous general anesthetics, such as propofol and etomidate, can produce severe burning pain on injection, presumably requiring the activation of nociceptive neurons. In addition, some commonly used inhaled anesthetics, such as isoflurane and desflurane, have a strong, pungent odor and can produce vigorous airway reflexes like cough and laryngospasm, which renders these anesthetics unsuitable for inhaled induction of anesthesia.
How do these observations fit with the general paradigm of anesthetics acting by reducing neuronal excitability and what are the molecular mechanisms of these adverse hyperalgesic and irritating effects? Anesthetics are thought to act on specific protein targets. and a few ion channel proteins, including the γ-aminobutyric acid subtype A (GABAA) and N-methyl-d-aspartate (NMDA) receptors, have emerged as the most likely candidates involved in producing loss of consciousness through the depression of neuronal excitability and synaptic transmission [for a comprehensive review, see (7)]. In addition to these direct effects on ion channels, anesthetics may also modulate neuronal signaling by affecting protein-protein interactions [for example, see (8)]. Nevertheless, the complex actions of general anesthetics are likely the result of effects on specific neuronal pathways (7). Much less is known about molecular mechanisms of undesired effects of general anesthetics such as airway irritation or injection pain.
Two exciting new studies provide evidence for a key role of TRPA1 and TRPV1, two members of the Transient Receptor Potential family of ion channels, in mediating excitatory side effects of general anesthetics in the respiratory system and the pain pathway (9, 10). C-fiber nociceptors mediate the responses of other known airway irritants, and TRPA1 and TRPV1 are activated by these irritants. There is now a growing list of irritant chemicals demonstrated to activate TRPA1, suggesting that it may function as a general chemosensor, activated by irritating and potentially dangerous chemicals, and as such, may be important in the chemoprotective mechanisms of vertebrates.
TRPA1 is expressed on a subset of primary sensory neurons that respond to capsaicin (11–14). TRPA1 has been implicated in cold sensation, hearing, and chemosensation for pungent chemicals (15). Its role in thermo- and mechano-transduction is questionable, but it has emerged as sensor for a wide variety of irritant chemicals, including the pungent ingredients of various plants, such as wasabi, mustard, and garlic (16). In addition, environmental irritants, such as acrolein and crotonaldehyde––both unsaturated aldehydes and constituents of cigarette smoke––also activate TRPA1 and contribute to cigarette smoke induced neurogenic inflammation (16, 17). Recently, 4-hydroxynonenal, which is generated by reactive oxygen species during inflammation, has been identified as endogenous agonist for TRPA1, inducing the release of calcitonin gene–related peptide and substance P to promote neurogenic inflammation and pain (18).
TRPV1 is the prototypical nociceptive ion channel and is activated by irritant chemicals, such as capsaicin and resiniferatoxin, as well as by noxious heat above 43°C and protons (19). Also, TRPV1 is essential to the thermal hyperalgesic response after tissue injury and inflammation (20). Given their role as chemosensors for environmental irritants, including pulmonary irritants, and their expression on peripheral nociceptors, both TRPA1 and TRPV1 appear strong candidates for mediating the irritant effects of inhaled anesthetics.
In the first of the two papers, Matta et al. (10) show that diverse irritant anesthetics activate TRPA1 heterologously expressed in HEK293 cells and endogenously expressed in dorsal root ganglion (DRG) neurons. The concentrations required for this activation span the clinical dose range for the irritant inhaled-anesthetics isoflurane and desflurane but are significantly above the plasma concentrations required to maintain propofol- or etomidate-mediated anesthesia. Local concentration peaks achieved during bolus injection of propofol or etomidate, however, are much higher––certainly in the range where TRPA1 activation has been demonstrated––and this mechanism may therefore be responsible for the burning pain on injection. Several experiments support these interpretations. The pungent inhaled anesthetics isoflurane and desflurane as well as the intravenous anesthetics propofol and etomidate activate TRPA1, as evident by inward currents and increases in intracellular calcium, in both cell systems (DRG neurons and HEK293 cells). In patch clamp experiments, neurons exhibited anesthetic-induced depolarization and action potential generation. The effects persist in electrophysiological experiments using excised membrane patches, arguing for a direct effect on the channel protein rather than a second messenger–mediated effect. In experiments testing for additive effects of anesthetics and alcohols on TRPA1, isoflurane did not produce any additive effect when applied together with an apparently saturating dose of octanol, whereas the effects of propofol and octanol were additive. The conclusion that isoflurane and alcohols activate TRPA1 through a common mechanism, distinct from the site of action for propofol, must be viewed cautiously because the propofol experiments were not done with saturating octanol concentrations and cannot, therefore, be directly compared. Behavioral experiments showed a TRPA1-dependent nocifensive behavior (a defensive or protective behavioral response, such as limb withdrawl, flinching, or licking) induced by propofol applied to the nasal epithelium. Furthermore, in a model of allyl isothiocyanate–induced neurogenic inflammation, the irritant anesthetic isoflurane augmented the inflammatory reaction more than the nonirritant sevoflurane.
Although TRPV1 and TRPM8, two other members of the TRP superfamily of ion channels, were not directly activated, the authors report in their second publication that anesthetics are capable of sensitizing TRPV1 (9). This study focuses on the effects of inhaled anesthetics on TRPV1 and shows that inhaled anesthetics, at clinically relevant concentrations, sensitize TRPV1 activation by heat, protons, and capsaicin. Pretreatment of cultured cells expressing TRPV1 (again, heterologous expression in HEK293 or endogenous expression in DRG neurons) with bradykinin (BK) or phorbol 12,13-di-butyrate (PDBu) [both leading to PKC activation] sensitizes TRPV1 to anesthetic activation. Activation of PKC through various pathways [for example BK acting through the BK type 2 receptor (B2R) to increase phospholipase C activity and, further downstream, PKCε activity] is one important mechanism in up-regulating TRPV1 and ultimately nociceptor sensitivity under conditions of inflammation (21) (Figure 1⇓). Some of the anesthetics tested are irritants (e.g., isoflurane and desflurane) and others can be classified as non-irritant (e.g., halothane and sevoflurane) and are clinically used for inhalation induction. Whereas the activation of TRPA1 correlated well with the irritant potential of the anesthetic (10, 22), the sensitization of TRPV1 did not correlate as well.
What is the possible clinical implication of these findings? Is there any evidence that nociceptor activation or sensitization by irritant anesthetics presents a clinical problem? Several animal studies support the affirmation of this hypothesis. For example, low concentrations of inhaled anesthetics decrease hind-paw withdrawal latency to painful heat in rats, indicating increased pain sensitivity or hyperalgesia (6). These findings have been confirmed in rats and mice by other investigators (3, 4, 23). Several mechanisms besides TRP channel activation have been proposed, including nicotinic and α-adrenergic pathways. Comparisons are difficult, however, because the models used differ substantially (3, 23–25). In human volunteers, nitrous oxide and methoxyflurane have analgesic properties at subanesthetic concentrations, whereas halothane, isoflurane, enflurane, and sevoflurane have no effect on thermal pain (5). Extensive studies of the analgesic effect of sub-anesthetic concentrations of isoflurane were conducted in human volunteers using a wide variety of experimental pain models and showed no clear analgesic effect (26, 27); however, none of the studies reports a hyperalgesic effect. Patients undergoing surgery with anesthesia from isoflurane (an irritant inhaled anesthetic) experienced more severe postoperative pain than patients who were anesthetized with propofol (28). Although a hyperalgesic effect of isoflurane is only one possible explanation for those findings that study emphasizes that the hyperalgesic effects may become only apparent under conditions of inflammation (i.e., surgery). To test this hypothesis, the effects of anesthetics with different irritant potential should be measured in models of inflammatory pain, ideally including a model of surgical pain.
Critics of this hypothesis will argue that given the widespread use of general anesthetics, including the irritant drugs shown to activate TRPA1 and sensitize TRPV1, a clinically relevant effect on postoperative pain could not have been missed. Nonetheless, several reasons exist for why this effect could be missed. Pain is considered a normal “side effect” of surgery by the patient, anesthesiologist, surgeon, and nursing staff, and variation in pain intensity among patients is usually assumed to be the cause of differences in extent of surgical trauma, degrees of inflammation, or in pain perception. The inflammatory response to surgical injury is well known, has been studied extensively, and its contribution to postsurgical pain is also well characterized. Varying degrees of inflammation obviously may be responsible for some of the clinically observed variability in postoperative pain intensity, making it difficult to recognize the adverse contribution of anesthetics. Last but not least, there likely has been a bias against this hypothesis: Why should a class of drugs that renders patients unresponsive to surgical stimulation act to worsen postoperative pain? Most studies testing the effects of general anesthetics on pain were therefore designed to detect analgesic and not hyperalgesic properties.
The irritant potential and adverse effects of anesthetics are more easily recognized in the airway. Volatile anesthetics have distinct odors and some (i.e., isoflurane and desflurane) are very irritating to the airways. Inhaled vapor anesthetics are also regarded as bronchodilators and are even sometimes suggested as therapeutics in the setting of status asthmaticus refractory to other interventions. This seems surprising in light of their irritant effects, which can trigger unpleasant sensations, such as cough, or broncho- or laryngospasm. Clinical evidence is not consistent, but it is clear that at least at certain stages of clinical anesthesia, the airway is much more irritable. This phenomenon is more pronounced in individuals with preexisting increased airway reactivity (e.g., recent respiratory infections, asthma, or current smoker). Published studies are quite complex and difficult to interpret since the methods used to study the effects of the anesthetics on the airway are not consistent. Several studies suggest that the inhaled anesthetics can reduce airway resistance at least temporarily. One study, using isolated and perfused rat lungs, found that all four inhaled anesthetics apparently reduced the increase in airway resistance produced by acetylcholine, but only desflurane and sevoflurane decreased basal airway tone (29). We have found that irritant inhaled anesthetics produce neurogenic bronchoconstriction mediated through nociceptors and likely requiring TRPA1 expression. This effect was blocked by pretreatment with neurokinin receptor antagonists, indicating that it involves a neurogenic inflammatory process that includes the release of neuropeptides [(22) and unpublished results]. This mechanism is distinct from the mechanisms proposed for bronchodilation which likely is mediated through direct effects on calcium homeostasis in smooth muscle cells (30). Depending on the concentration of anesthetic and any preexisting inflammation (asthma, tobacco, etc.) the net effect will be constriction or relaxation.
Should we change our clinical management based on these findings? It is too early to recommend a general change in clinical practice favoring non-irritant over irritant anesthetics. The results of these basic research experiments are convincing, but more studies are needed to determine if there is a clinically relevant difference between these different groups of anesthetics. However, there is more evidence accumulating that “anesthesia matters” (28). The choice of anesthesia may be important for the intensity of postoperative pain. With respect to preexisting airway disease, non-irritant anesthetics may be the better choice for avoiding further inflammatory insult.
General anesthetics, including irritant drugs, have been shown to have beneficial effects as well, such as preconditioning of myocardium (31), which protects it from ischemia, and possible neuroprotective effects (32).
It is fair to say that inhaled anesthetics have long been regarded as inert substances for which inhalation and exhalation determine the on- and offset of drug effect. More and more evidence has been accumulating that anesthetics have effects well beyond the end of surgery. These effects, when better understood, will likely have a significant impact on the choice of anesthetic.
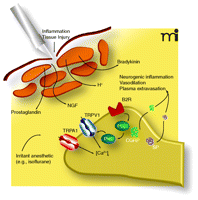
Mechanisms of sensitization (simplified) in peripheral nociceptors after tissue injury and inflammation and proposed interaction of inhaled anesthetics. Tissue injury leads to activation of nociceptive nerve terminals, which will then trigger the release of the neuropeptides calcitonin-gene-related-peptide (CGRP) and substance P (SP). CGRP causes arteriolar vasodilatation and SP induces a vascular leak leading to plasma extravasation. Tissue injury will further lead to release of inflammatory mediators including cytokines, proteases, bradykinin (BK), nerve growth factor (NGF), histamine and 5-hydroxytryptamine (5-HT). Together, these factors act to increase the sensitivity of the nociceptor by different specific mechanisms. Some of these mechanisms converge and lead to activation of protein kinase C and to increase in intracellular calcium, an important second messenger molecule. Through activation of TRPA1 and sensitization of TRPV1, certain irritant general anesthetics can potentially augment this sensitization or may even lead to clinically relevant sensitization independent of injury. Together, these effects might contribute to postoperative pain and airway irritation. PLC, phospholipase C; NGF, nerve growth factor.
Acknowledgments
I would like to thank Nigel Bunnett, Mark Schumacher, and Pamela Derish for helpful comments on the manuscript. This work is supported by UCSF and the Hellman Family Foundation.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Helge Eilers, MD, is an Associate Professor in the Department of Anesthesia and Perioperative Care, University of California, San Francisco. He received his MD at the University of Bonn, Germany and completed his anesthesia residency training at UCSF. His research focuses on the study of the molecular biology and physiology of pain transduction in the peripheral nociceptor. E-mail eil-ersh{at}anesthesia.ucsf.edu; fax 415-514-0185.

