Advanced Drug Delivery Systems That Target The Vascular Endothelium
Abstract
Targeted drug delivery to endothelial cells lining the vascular lumen will provide effective, precise andsafe therapeutic interventions for treatment of diverse disease conditions. Rational design of such drug delivery systems (DDS) includes the following intertwined tasks: 1) selection of proper target determinants on endothelial surfaces, such as cell adhesion molecules, ectopeptidases, or caveolar antigens; 2) production of affinity ligands useful for targeting, such as affinity peptides, antibodies, or their fragments; 3) selection and adopting of suitable delivery vehicles (such as liposomes or polymer nanocarriers); and 4) formulation of DDS with optimal targeting and therapeutic features. Important therapeutic features of DDS include: 1) sufficient targeting effectiveness, circulation time, and safety (i.e., lack of systemic and local adverse effects); 2) precise subcellular localization of drugs targeted to endothelial cells; and 3) adequate amplitude, kinetics, and duration of effects. This review utilizes examples of DDS-mediated interventions in vascular inflammation, oxidative stress, and thrombosis and analyzes them in an attempt to create design parameters that best regulate the pharmacological and therapeutic features of DDS that target endothelial cells.

Introduction
A monolayer of endothelial cells (EC) that lines the luminal surface of blood vessels controls vascular tone, blood fluidity, and extravasation of blood components (1). Endothelium is a specialized tissue that consists of diverse EC subtypes (2) and plays a central role in ischemia, thrombosis, inflammation, and vascular oxidative stress––all morbid syndromes involved in pathogenesis of stroke, ischemic heart disease, acute lung injury, atherosclerosis, hypertension, and diabetes (3). Thus, the endothelium represents a key target for pharmacological interventions in a plethora of disease conditions, including cardiovascular, hematological, pulmonary, metabolic, oncological, and genetic maladies. However, because of their lack of affinity to the endothelium, only a small fraction of injected therapeutics binds to EC. An additional challenge is that many drugs require precise delivery to specific cellular compartments. The goal of endothelial targeting is to achieve specific and safe delivery of a drug to, into, or across EC, in order to localize effects in the lumen, desired intracel-lular EC compartments, or subendothelial space, thereby improving pharmacological interventions (4, 5) (Box 1).
Endothelium: A Key Vascular Target
A couple decades ago, the monolayers of endothe-lial cells lining the luminal surfaces of blood vessels were viewed as a passive “carpet,” helping to keep platelets inactive and minimizing unintended coagulation. Our increased understanding of the integral functions of endothelium in governing vascular tone and the blood-tissue interface has had the effect of placing endothelium on the “short list” of the most important therapeutic targets for molecular interventions. It also became clear recently that endothelial cells (EC) in the vascular system represent diverse subpopulations, each of which possess characteristic determinants that can be utilized for targeting drugs. Today, design of Drug Delivery Systems (DDS) targeting to EC is an exponentially growing field of biomedical research aimed at optimizing the treatment of cardiovascular and many other pathologies, including metabolic and oncological diseases.
This review offers a brief discussion of several key biotech-nological, molecular, and cellular considerations important in the context of designing drug delivery systems (DDS) targeted to EC. We focus on biotherapeutics (such as therapeutic enzymes), but in order to avoid overt superficiality, we do not discuss the delivery of nucleic acids. We also consider detailed examples of systems directed to EC determinants (i.e., proteins expressed on the surface of EC) that have possible utility for therapeutic interventions aimed at the management of vascular pathological processes.
Designing DDS that target EC is a complex pursuit of several intertwined tasks (Figure 1⇓). Because of space limitations, this article gives rather a cursory overview of many aspects of this paradigm. The aspects related to selection of target determinants and affinity moieties (Figure 1, I and II⇓) have been discussed in detail in recent reviews (4–6). Formulations of nanocarriers and their pharmacological properties (Figure 1, III and IV⇓) have been reviewed elsewhere (7). Aspects related to subcellular addressing of DDS and potential side effects in endothelium (Figure 1, IV⇓) have been reviewed in details in recent reviews and chapters (5, 6, 8).
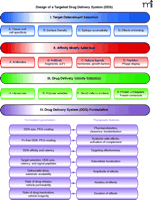
Targeted drug delivery systems (DDS) design. This schematic flowchart illustrates a generic algorithm for rational design and key parameters of a targeted drug delivery system. This review analyzes aspects of this schema that are pertinent to DDS targeting to endothelial cells (EC). For the sake of coordinating diverse parameters, the schema depicts a sequential organization of DDS design, divided into four blocks, from target selection to characterization of its pharmacokinetic and therapeutic features. Each block is divided into a few major parameters or categories. In practice, a DDS design may start with the synthesis of a new drug vehicle potentially suitable for EC targeting or the selection of a peptide that binds to EC or certain subtypes of EC. Final therapeutic features of a given DDS are governed by a complex interplay between factors depicted in all four blocks. For example, circulation and systemic side-effects of a DDS are dependent on the features of both affinity moiety (e.g., Fc fragment–mediated clearance and side effects) and vehicle (e.g., presence of PEG-coating providing stealth features), whereas subcellular localization of a drug is controlled by features of the selected target determinant, valency, and size of DDS.
Selection Of Target Determinants
Selection of an optimal molecular determinant on the EC surface that can be used to anchor drugs is the first logical step in design of a targeted DDS (Figure 1, I⇑). Modern high-throughput techniques have helped identify such determinants, including endothelial ecto-peptidases (9, 10), caveolar proteins (11), cell adhesion molecules (6, 12, 13) receptors for growth factors and transport proteins (14, 15) and other proteins [reviewed in (5)].
For example, EC plasmalemma proteomics characterizes the molecular topography of surface determinants throughout EC in organs of interest and identifies “zip codes” characteristic of vascular areas (16, 17). In addition, separation methods allowing proteomic analysis of specific domains of EC plasmalemma, such as caveoli (18), permit micro- and nano-scale range “cartography” of EC surfaces.
Repetitive cycles of intravenous injections of phage display libraries encoding stochastic peptides that may bind to accessible sites in the vasculature permits the identification of peptides homing into selected tissues, by sequencing the DNA of the phage isolated from an organ of interest, and eventually, by isolating the binding sites (19, 20). The advantage of this method lies in the built-in recognition (and selection) of sites accessible only to the blood (21).
Criteria That Define A Good Target
A good target determinant must meet at least four criteria if it is to be considered worthy of further investigation. First, it must be expressed on EC and its presence in blood or other cell types accessible to blood should be low, otherwise, counterparts of the target determinant circulating in blood [such as P-selectin that is expressed on both EC and platelets (13)], may compromise delivery to the EC. Absolute specificity, however, is not necessary; targeting to EC will suffice if a competing determinant is localized in a counterpart cell type inaccessible to blood (9).
Second, surface density of the target must be sufficient to promote the binding of the effective therapeutic doses of drugs. For example, certain EC adhesion molecules, such as CD31 (PECAM-1), are present in multi-millions of copies on the surface of each cell (6), and angiotensin-converting enzyme (ACE) is constitutively expressed at hundreds of thousands of molecules per cell (22). Additionally, pathological factors can affect the expression of EC surface determinants: the expression of intercel-lular adhesion molecule-1 (ICAM-1) and other adhesion molecules (such as selectins) is greatly enhanced in conditions of inflammation, thrombosis, and ischemia (23–25).
Third, a binding site must be accessible to blood. Intracellular components are not useful for drug delivery, unless they become exposed on the lumen under pathological conditions (e.g., selec-tins). Steric accessibility of the binding site(s) is necessary for association with circulating carriers, which range in size from small fusion proteins to carriers 100–300 nm in diameter (see below). In cases of determinants transiently expressed under pathological conditions such as EC selec-tins (26, 27), the identified time-window of their exposure to the bloodstream should suffice for targeting.
Fourth, the consequences of DDS binding to a target determinant must meet therapeutic goals. Thus, targeting should not cause harmful side effects to EC. Binding of drugs may activate shedding and internalization of target molecule, or otherwise inhibit or activate them, which may lead to side effects, such as signaling and EC activation. Ideally, binding of an antibody-drug complex to a target antigen should cause additional therapeutically beneficial side-effects. For example, blocking ACE activity (28) or cell adhesion molecules (29, 30) on EC may be beneficial in the context of hypertension or inflammation, respectively. Furthermore, docking to a target molecule should provide a proper subcellular localization of a drug. For instance, EC determinants localized in caveoli (flask-shaped invaginations of EC plasma membrane with diameter 70–80 nm) favor intracellular and trans-endothelial delivery (18, 31). The EC cell adhesion molecules PECAM-1 and ICAM-1 offer an array of subcellular delivery sites (6), based on the rational design of DDS.
Affinity Moiety Selection
There are many classes of molecules that can serve as affinity ligands for EC targeting (Figure 1, II⇑). Natural ligands of endothelial receptors, such as transferrin and growth factors including VEGF (vascular endothelial growth factor), can be conjugated with drugs or drug carriers to facilitate binding to and entry into EC (14, 32). In some cases, selection of the targeting ligands steams from the methodology of identification of target determinant, such as phage display libraries providing affinity peptides or single-chain antibody fragments binding to EC (19, 20, 33).
Currently, immunoglobulin G (IgG)-type antibodies represent arguably the most popular class of affinity ligands for targeting, because they are well suited for conjugation with drugs and drug carriers. Modular domain structure of IgG consisting of Fab- and Fc-fragments (i.e., affinity and accessory moieties, respectively) permits rational bioconjugation and control of valency (i.e., the number of binding sites) and size of affinity ligands such as bivalent 160-kD IgG, bivalent 120-kD F(ab)2, and monovalent 60-kD Fab. Elimination of the Fc-fragment circumvents potential side effects that arise from the activation of complement and leukocytes. Valency and size are among key parameters affecting pharmacokinetics, targeting, local side effects, and sub-cellular addressing of DDS (Figure 1, IV⇑).
Recombinant techniques enable modular molecular engineering of monoclonal antibody fragments, such as the production of a single-chain Fv (scFv), the minimal functionally active monovalent antigen-binding fragment of IgG that consists of a VH domain and a VL domain. Multivalent scFv iterations linked by short connecting peptides are also possible and provide multiple binding sites that recognize targets but carry a minimum of unnecessary antibody peptide sequences, thus reducing the possibility of unintended side-effects. These small antigen-binding species provide an ideal format for synthesis of Fc-fragment free conjugates of therapeutic agents (e.g., enzymes, cytokines) with controlled size and valency, using chemical or recombinant fusion conjugation methods.
Technologies for producing homogeneous, Good Manufacturing Practices (GMP)-amenable monoclonal antibodies and their fragments have been developed and accepted by the biotechnology and pharmaceutical industries, which saw exponential growth of FDA-approved antibodies and antibody-based pharmacological formulations in the last decade. Recombinant techniques have been developed for the grafting of murine antigen-binding fragments into human IgG (humanization), replacement of immunogenic amino acids in IgG (de-immunization), and enhancement of affinity by point-mutagenesis (antibody maturation), which further enhance the safety and specificity of antibody applications in human patients.
Drug Delivery Systems (DDS) For Vascular Targeting
Drugs can be loaded into liposome or polymer vehicles conjugated with affinity moieties (Figure 1, III⇑). Other available types of vehicles include blood proteins or erythrocytes (which are not discussed in this review). Alternatively, drugs can be directly conjugated with antibodies or other affinity moieties. For example, antibiotics, toxins, cytokines, and enzymes have been conjugated with antibodies or antibody fragments. Recombinant fusion proteins that consist of an effector part (e.g., enzyme) and a targeting fusion protein represent a specific example of direct conjugates, which also will be discussed below in the context of targeting EC.
Recent research has focused on the design of liposome and polymer-based nanocarriers for drug targeting to EC. Functions of nanocarriers include: 1) optimization of a drug’s pharmacokinet-ics in the bloodstream and protection of drugs against inactivation and premature activity en route to the target; 2) fine control of drug-release kinetics; 3) providing a template for multivalent affinity binding sites enhancing effectiveness of anchoring on the target cells; and 4) modulation of subcellular delivery of drugs. Nanocarriers are broadly defined as submicron structures that can be loaded with drugs (34). Key controllable parameters of nanocarriers that define their utility for drug delivery include structural materials, plasticity, morphology, size, shape, permeability, biocompatibility, and biodegradability. Figure 2⇓ illustrates the structural design of some currently available nanocarriers.
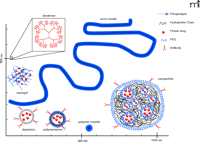
Nanocarriers for vascular drug delivery. Schematic representation of several main types of nanocarriers depicted in relative size scale with limited and oversimplified structural features. All nanocarriers can be surface-conjugated with targeting antibodies (or alternative affinity moieties) and PEG polymer providing stealth features. Dendrimers are the smallest of nanocarriers, in the maximum range of tens of nanometers. They possess multiple end groups suitable for high extent of coupling targeting or active agents. Liposomes, composed of biologically derived phospholipids, are the most common form of nanocar-riers with large aqueous loading potential, yet are also the least stable of carriers. Polymersomes, one of three self-assembled polymeric nanocarriers, are the synthetic polymer analog of liposomes, possessing enhanced stability, dense PEG coating, and therefore prolonged circulation in vivo. Worm micelles are long, flexible cylindrical polymer micelles which possess one of the longest circulation times recorded in vivo. Polymeric micelles are the smallest of the self-assembling polymer aggregate carriers and are the least stable self-assembler. Nanogels are composed of cross-linked polymers with drug entrapped into the ensuing matrix. Their circulation and potential therapeutic uses still remain to be studied. Nanoparticles are solid polymer structures formed through processing rather than self-assembly methods. They represent the largest of the carriers, have the greatest active protein loading capacity measured to date, and are able to protect encapsulated therapeutic enzymes from external proteolysis.
Phospholipid-based liposomes arguably represent the most extensively studied drug vehicles (35, 36). Phospholipids forming bilayers in aqueous media provide capsular vehicles, the internal aqueous space of which can be used for delivery of hydrophilic drugs while the lipid bilayer can be loaded with small hydrophobic drugs. Liposomes can be made within a homogeneous and narrow size distribution: 100–200 nm diameter liposomes are most suitable of vascular route.
Liposomes and other vehicles are cleared from the bloodstream within a few minutes, owing to the binding of plasma complement, immunoglobulins, and other proteins (opsonization), leading to uptake by macrophages in the reticuloendothelial system (RES, including liver and spleen) (35). This clearance can be markedly delayed by the grafting of synthetic hydrophilic polymers such as poly(ethylene glycol) (PEG) onto the surface of the vehicles. PEG forms a hydrated shell hindering protein interaction with drug vehicles or drugs themselves (e.g., PEG-coated insulin), thereby greatly reducing opsonization and uptake by macrophages. Such “stealth” DDS have prolonged pharmacokinetics and lesser side effects of activation of host defense (immune response, cytokine release, complement activation) (35).
The extent of stealth effects depends on PEG surface density: in theory, grafting 100% of the phospholipid surface of liposomes would provide better stealth features. Alas, liposomes can only contain ~ 15 mol% PEG because a greater extent of grafting hydrophilic PEG destroys the phospholipid bilayer (37, 38). Polymersomes, the polymer analog of liposomes, are free of this limitation. Each polymer chain in polymersomes can contain a PEG group, hence 100 mol% surface coverage is possible, resulting in circulation half-life of days as compared to hours for PEG-liposomes. Additionally, the membrane of polymersomes is thicker than that of liposomes (~8 nm compared to ~3 nm), providing highly durable carriers that are able to resist deforming forces that destroy liposomes (37, 38).
The copolymers used for polymersome synthesis include PEG-conjugated poly(lactic co-glycolic acid) (PLGA), poly(butadiene), and poly(ethylethylene) (38). Internal aqueous domain of polymer-somes can be loaded by hydrophilic drugs with sizes up to 500 kD, although the encapsulation efficiency decreases with increasing molecular mass (39). This dependence of loading on drug size is a general feature of all nanocarriers that use natural partitioning for loading. Encapsulation of small solutes (e.g., sucrose and glucose) is generally effective at high concentrations and, therefore, limited practically by their solubility; however, harsh encapsulation conditions, required to overcome the more durable nature of polymersomes (e.g., high temperature), are more likely to affect the stability and activity of sensitive bioactive agents such as therapeutic enzymes.
By changing the ratio between the hydrophobic and hydrophilic polymer parts (or blocks), in a copolymer (e.g., formed at the relative PEG content in PEG-PLGA co-polymer at range of ~ 42–50%), the polymer chains can self-assemble into cylindrical, flexible structures known as worm micelles, with widths of ~ 40 nm and length up to 20–40 μm (37, 38, 40). These species––which served for decades as a laboratory curiosity––have been only recently conceived as a new prospective class of drug carriers. A unique and highly attractive feature of worm micelles is their ability to align with flow (41), which presumably may enhance circulation even further by avoiding collisions with vascular cells. Worm micelles have no internal aqueous space and thus cannot encapsulate hydrophilic drugs. Hydrophobic and amphiphilic drugs can be intercalated into the hydrophobic polymer core, however, while hydrophilic drugs can be coupled to the surface of the worm micelles.
Polymer micelles, formed when the PEG content in the di-block copolymer (i.e., consisting of two polymers) is >50% (37), can be used for loading of poorly soluble hydrophobic drugs. Their thermodynamically driven self-assembly dictates uniformity in size and shape in the spherical core. However, in contrast with other polymer-based nanocarriers, which release encapsulated drugs via relatively slow surface erosion process, polymer micelles release their load via burst release, with nearly 80% of cargo lost within the first six hours (42). Recently, polymer pro-drugs have been synthesized by conjugating the drug directly to the hydrophobic block of the carrier polymer, hence drug release is dictated by the degradation of the polymer and not the diffusion of the drug out of the carrier (43, 44).
Liposomes, polymersomes, worm micelles, and polymer micelles are formed by a thermodynamically driven self assembly process dictated by mixing of hydrophilic and hydrophobic components. In contrast, nanoparticles are machined structures whose shape, size, and drug loading are dictated almost entirely by the processing conditions used. Once formed, they are the most stable of the nanocarriers (7). Further, upon injection into the bloodstream, they are not subject to the same destabilizing effects of plasma components that shorten half-life of many self-assembled carrier structures.
Nanoparticles can be produced by methods including homogenization of single and double emulsions, gas expansion, and solvent extraction (42). Depending on the materials and synthesis method, the size of polymer nanoparticles may vary from 50 nm up to 400–600 nm. Currently available methods yield formulations with a wide distribution in particle size; however, rational selection of the surfactants and homogenization conditions used in synthesis can also direct the formation of special nanoparticle geometries (e.g., ellipsoids). Hydrophobic agents can be incorporated into the polymer matrix during the formulation.
Hydrophilic drugs can be loaded into internal aqueous caverns during nanoparticle formulation or conjugated to the particle surface afterwards; the latter approach is used for coupling affinity moieties. Previously tested conditions for drug encapsulation during the nanoparticle formulation process led to the inactivation of heat-sensitive drugs and enzymes, but new methodologies have created polymer nanoparticles encapsulated with significant amounts of active enzymes (e.g., 15–20% yield encapsulation of catalase), which are protected from the proteolytic environment by the polymer shell (45).
Dendrimers (46), the smallest of nanocarriers––a few nano-meters in diameter, represent polymer chains branching at regular bifurcating intervals, providing rapid expansion in the number of end groups with increasing molecular mass. Dendrimers are typically very uniform in size, and can achieve very high molecular masses beyond 1,000 kD. Because of their highly branched structure, they tend to adopt a spherical geometry that provides dendrimers with a very low inherent viscosity, as compared to linear polymers of the same molecular weight. Owing to their small size, dendrimers have very high surface-to-volume ratio and serve as effective clustering domains for the formation of drug aggregates.
All the carriers mentioned so far have used copolymers with hydrophobic domains that form the core structure of the carrier, hindering encapsulation and activity of hydrophilic drugs, especially proteins. Nanogels (47), based on cross-linked hydrophilic materials forming insoluble gels that swell in water, may bypass these limitations. The crosslinks of polymer networks can be covalent bonds, entanglements, ionic interactions, or affinity-ligand pairings. Because of the hydrophilic nature of the inner volume and structural components of nanogels, loaded proteins and other hydrophilic drugs remain in a native conformation; nonetheless, uncontrolled aggregation is a technical challenge for formulation of injectible nanogels.
The main requirement for any nanocarrier material is biocom-patibility, which means they can be injected intravenously without toxicity and overt side-effects including activation of leukocytes, platelets, complement, coagulation, and kinins (48). Clearance mechanisms for inert materials in the nanoscale range (e.g., elimination via urine or bile excretion), however, remain to be characterized; hence, all materials used for nanoparticles must be both biocompat-ible and biodegradable. At the very least, these materials should degrade into soluble components that are ≤ 50 kDa in size and are non-toxic. For example, PLGA degrades into lactic and glycolic acid residues under hydrolysis conditions, providing easily metabolized and excreted degradation products (Box 2).
Drug Delivery Systems (DDS) for Vascular Targeting
Diverse synthetic (e.g., liposomes, polymer nanocar-riers) and natural (e.g., blood cells and proteins) objects can serve as vehicles for vascular drug delivery. They range in size from nanometers to micrometers, help to optimize the pharmacokinet-ics of a drug, and isolate the drug from the body en route to the therapeutic target. Endothelial cells represent a good target; they are highly accessible to relatively large drug vehicles circulating in the blood. Coating the drug vehicles with the hydrophilic polymer poly(ethylene glycol) (PEG) markedly prolongs their circulation time and reduces potentially dangerous side effects such as the activation of complement and phagocytes. Antibodies and other molecules (that possess affinity to endothelial determinants) are conjugated to the vehicles and provide specific targeting. Molecular modifications of these affinity moieties (e.g., synthesis of recombinant small antigen-binding fragments), provide modular templates for the design of DDS with minimal side effects and controlled valency of binding to their targets.
Protein Conjugates
Protein conjugation [e.g., covalent coupling of an antibody to an enzyme (49, 50)], can also provide nanoscale DDS (Figure 1, II⇑). Proteins provide diverse reactive groups for conjugation, such as sulfhydryls of cysteine, primary amines of lysine and N-termini, as well as carboxyl groups, etc. Furthermore, additional reactive groups may be chemically introduced in the protein using activating reagents. For example, a maleimide group, which is highly reactive to sulfhydryls, can be introduced into one protein via an amino group by using succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) and will react with SH-groups of another protein. The methods of protein conjugation are well established (50).
Series of immunoconjugates of anti-thrombotic and antioxidant enzymes for targeting to EC, have been designed using biotin-streptavidin and chemical conjugation (49, 51, 52). Advantages of immunoconjugates for the targeting of enzymes to EC include high conjugation efficiency and a wide range of controllable size of resultant conjugates, although the shape and size of large conjugates are difficult to control. Thus, an antibody-enzyme conjugate may consist of one molecule of antibody linked to one molecule of a drug or may consist of multiple copies of each molecule and reach size of 100–500 nm––larger conjugates may precipitate during reaction. In contrast to liposomes or nanocarriers, the size of a protein conjugate depends on the extent of modification of both components and is not determined thermodynamically (49) and may result in relatively higher heterogeneity of the product (52). Protein immunoconjugates are an easily biodegraded DDS.
The targeting ability and the effects of diverse therapeutic enzymes directly conjugated with ACE-specific, PECAM-specific, or ICAM-1-specific immunoglobulins have been tested in cell cultures and in animal studies. When the antioxidant enzyme catalase was conjugated to these antibodies and bound determinants on EC, the EC were protected against damage by H2O2 in vitro, in perfused organs, and in intact animals (10, 51, 53–55). Additionally, anti-thrombotic plasminogen activator (tPA) conjugated with ICAM-specific immunoglobulin demonstrated highly efficient targeting to the endothelium in mice (56). Recombinant fusion of enzymes (or their active domains) with antigen-binding fragments of antibodies directed to EC represent yet another, highly advantageous drug delivery system.
Subcellular Delivery Of DDS To Endothelial Cells
The intracellular destination is a key parameter determining the effects, metabolism, and lifetime of the delivered agents (8, 57). For example, intracellular delivery of antioxidant enzymes (e.g., catalase) helps to detoxify oxidants otherwise poorly accessible for circulating drugs. Endocytic pathways, naturally useful for intracellular drug delivery, include: 1) macropinocytosis and phagocytosis, main pathways in macrophages allowing uptake of extracellular fluid or large particulate ligands, respectively (6, 58–60); 2) classical clathrin- and caveolar-mediated endocytosis, used by most cell types, including EC, for internalization of diverse ligands and turnover of plasma-lemma components (11, 61); and 3) recently defined endothelial CAM-mediated endocytosis, described in more detail below.
The internalization pathway determines the final destination of the internalized materials. Macropinocytosis, phagocytosis and clath-rin-mediated pathways generally deliver materials via endosomes to lysosomes (62, 63), whereas caveoli-related endocytosis delivers materials to a wider list of compartments including the cytosol, the Golgi complex, and the endoplasmic reticulum, in addition to lyso-somes (64, 65). Endocytic vesicles internalized via clathrin-mediated and, more typically, caveolar-mediated endocytosis can also traverse the cytosol, thus transporting materials across EC (65, 66).
Targeting nanoconjugates to EC receptors that are naturally associated with specific endocytic pathways often leads to internalization and intracellular trafficking typical of these pathways (8). For example, liposomes and other DDS targeted to E- or P-selectin––proteins whose turnover in EC naturally occurs via clathrin-mediated endocytosis (67, 68)––enter EC via clathrin-coated pits and traffic to lysosomes (27, 69). Imaging probes and drugs conjugated to transferrin receptor–specific antibodies bind to this receptor, which is expressed on the luminal surface of brain EC, enter EC via clathrin-coated pits, and traverse EC, similarly to the endogenous ligand transferrin, thus supporting drug delivery through the blood-brain-barrier [reviewed in (70)]. DDS that exploit caveolar-mediated endocytosis (e.g., compounds conjugated to antibodies to gp90, a 90 kDa glycoprotein located in the caveoli in pulmonary endothelium) also provide transcellular delivery across EC (18).
Our present understanding, however, of the internalization mechanisms and subsequent trafficking pathways of nanoconjugates targeted to EC is incomplete. For instance, vascular cell adhesion molecule-1 (VCAM-1) is normally internalized from EC surface and transported to lysosomes via clathrin-mediated endocytosis (71). VCAM-1-specific antibodies have been tested for targeting of imaging agents or biodegradable carrier particles to EC altered by pathological factors in vitro and in vivo (72–74), but the itinerary of intracellular trafficking and final destination of VCAM-specific nano-conjugates remain to be determined; hence, the therapeutic utility of this drug delivery system remains unclear.
Intracellular delivery can also be achieved utilizing protein transduction domains (PTDs), such as those derived from the HIV transcription factor Tat (75, 76). The basic charges on Tat mediate binding to the negatively charged components of the plasmalemma, facilitating Tat’s uptake into the cells (75–77), which may occur via passive endocytosis of surface-bound Tat carriers or other, still elusive, mechanisms (75).
Targeting EC surface determinants other than endocytic receptors can also provide intracellular delivery of DDS via induction of endocytic processes that normally are not utilized by target cells. The targeting of DDS to the transmembrane glycoproteins ICAM-1 and PECAM-1, which are primarily expressed by EC, illustrate this scenario (25). Docking of natural ligands (e.g., binding of counter-proteins expressed on the surface of leukocytes) to ICAM-1 or PECAM-1 does not prompt uptake by endocytosis but results in proteolytic shedding of these molecules from the plasmalemma and ligand release (78, 79). Similarly, monomeric ICAM-1-specific or PECAM-1-specific antibodies are not internalized by EC (51, 52, 56). However, when these antibodies bind to cells as multivalent complexes (e.g., as multimeric protein conjugates or nanocarriers coated with “CAM”-specific antibodies), they cluster their specific antigens on the EC surface and enter EC very rapidly (t1/2 = 15 min) and efficiently (90% internalization of the cell-bound DDS) (52, 81). Interestingly, EC internalize protein CAM-specific DDS in the size range of 100–500 nm, but not very large (μm size) polymorphous DDS possessing the same content (52).
The EC-regulated internalization of CAM-specific DDS requires the activity of dynamin, a large GTPase that is typically involved in clath-rin- and caveolar-mediated endocytosis; however, this process of internalization does not utilize clathrin or caveolae or other previously known endocytic pathways (80). In fact, the uptake and subsequent intracellular trafficking of ICAM- or PECAM-specific DDS by EC (CAM-mediated endocytosis) has unique features (Figure 3⇓), including the activation of protein kinase C (PKC), Src (and related tyrosine kinases), and the Rho-dependent kinase (ROCK) pathways and formation of actin stress fibers (57, 81). ICAM-1 clustered by CAM-specific DDS interacts with the sodium-proton exchanger protein NHE1, which may serve as an adaptor for cytoskeletal proteins α-actinin and ERM (ezrin, radixin, and moesin) family proteins, forming actin filaments to the plasmalemma binding sites. Cytoskeletal reorganization and dynamin activity induce invaginations that pinch off the plasma membrane, driving ICAM-1-specific DDS and, concomitantly, ICAM-1 and NHE1, into vesicular compartments in the cytosol (25, 57, 81).
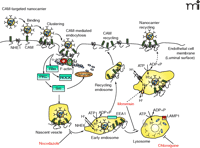
Intracellular drug delivery into vascular endothelial cells using CAM-specific DDS. Multivalent CAM-specific DDS bind to and cluster cell adhesion molecules (CAMs). This action activates protein kinase C (PKC), Src (or other protein tyrosine kinases), and Rho-dependent kinase (ROCK), which regulate the activity of the GTPase dynamin and the amiloride-sensitive sodium/proton exchanger NHE1, all of which induces the formation of actin stress fibers. CAM and NHE1 form a complex, which may help crosslink actin fibers to the CAM cytosolic tail through α-actinin (α-act) and ezrin/radixin/moesin (ERM) family proteins. After internalization, CAM-specific DDS traffic via a microtubule-dependent pathway to endosomes (a process that takes 1–2 hrs) that can be identified by the presence of the protein marker early endosome antigen-1 (EEA1). Endosomes are enriched in NHE6, an intracellular ion exchanger that helps regulate endo-some acidification through the vacuolar H+/ATPase. CAM-specific DDS dissociate from CAM and NHE1 in endosomes and CAM recycles to the plasma membrane. DDS arrive to lysosomes about three hours after their internalization; this process can be identified by colocalization with lysosome-associated membrane protein 1 (LAMP1). In lysosomes, NHE6 becomes inactive, favoring further acidification and degradation of delivered proteins by acidic proteases. Nocodazole (which disrupts the cell microtubule network), chloroquine (a mild base that inhibits lysosomal acidification), and monen-sin (which enhances Na+/H+ exchange in endosomes and induces recycling of CAM-specific DDS to the plasma membrane) can alter the intracellular itinerary of CAM-specific DDS and prolong their effects.
CAM-mediated endocytosis, taken with other features of targeting ICAM-1 and PECAM-1, provides a sound basis for intracellular drug delivery. For instance, CAM-targeted delivery of catalase by protein conjugates or polymer nanocarriers confers antioxidant protection to EC in models of oxidative stress in vitro and in animals (25, 54, 55, 80).
One hour after uptake, ICAM-1-specific DDS traffic to early endosomes (Figure 3⇑), which contain early endosome antigen 1 (EEA1, an endosome compartment marker) and an intracellular NHE isoform, NHE6 (57). NHE6 regulates ion balance and maintains a moderately low pH in the endosome, required for ligand dissociation from receptor and lysosomal biogenesis (82, 83). Indeed, ICAM-1-specific DDS dissociate from ICAM-1 within this compartment, and ICAM-1 recycles back to the plasma membrane, while DDS arrive to lysosomes about three hours after internalization (25, 80). Typically, ligands internalized via other pathways (e.g., clathrin-mediated mechanisms, macropinocytosis, and phagocytosis) are delivered to lysosomes within minutes (59, 84). The relatively slow traffic of CAM-specific DDS to lysosomes permits prolonged activity of targeted therapeutics, such as several hours of antioxidant protection by delivered catalase (80). However, NHE6 becomes inactive within lysosomes, and the pH drops because of unopposed activity of H+-ATPase (85), which activates lysosomal acidic hydrolases and results in proteolytic degradation of the delivered proteins (Figure 3⇑). Hence, the effect of catalase delivered intracellularly by CAM-specific DDS decays about three hours after internalization (80).
Understanding the interactions between CAM-specific DDS and the EC has permitted the optimization of “cellular pharmaco-kinetics” of these DDS (Figure 3⇑). For instance, the NHE1 inhibitor amiloride delays internalization of CAM-specific DDS by EC (81), which may suit therapeutic interventions intended for vascular lumen (e.g., fibrinolytics) (56). Additionally, disruption of the cell microtubular network by nocodazole inhibits lysosomal trafficking of CAM-specific DDS, whereas inhibition of lysosomal acidification by the mild base chloroquine protects delivered proteins against proteolysis. Monensin, which enhances Na+/H+ exchange in early endosomes, favors the sorting of DDS to a recycling pathway (57). All these pharmacological agents markedly prolonged the antioxi-dant effect of CAM-specific DDS of catalase (57, 80). Furthermore, ICAM-1 recycling supports recurrent targeting of subsequent doses of CAM-specific DDS because a single target molecule can uptake many rounds of circulating DDS, enhancing the efficacy of delivery (25). Therefore, auxiliary pharmacological interventions may help modulate the subcellular delivery, longevity, and effects of therapeutics targeted to EC by targeted DDS.
Finally, lysosomal destination of CAM-specific DDS can be viewed as an advantage in the context of certain therapeutic interventions, such as enzyme replacement therapies for lysosomal storage disorders––a group of syndromes primarily caused by inherited deficiency of lysosomal hydrolases (86). Indeed, CAM-specific DDS provide a means to bypass obstacles encountered by classical lyso-somal enzyme replacement therapies (e.g., poor targeting and endo-cytosis of recombinant enzymes) and markedly enhance effective enzyme delivery to EC lysosomes (87) (Box 3).
Subcellular Localization of DDS Targeted to Endothelium
The effects of most drugs depend on their subcel-lular localization. For example, many anti-thrombotic drugs should reside on endothelial surfaces, whereas drugs correcting lysosomal defects should be delivered intracellularly. The precise subcellular addressing of targeted drugs can be achieved by proper selection of endothelial surface determinants, which can either internalize bound drugs into cells through endocytic pathways or provide prolonged residence on the surface. Size and valency of a DDS binding to the endothelium can modulate the process of internalization. Thus, multivalent binding to certain constitutively expressed endothelial determinants that are normally not internalized, can lead to the clustering together of the EC’s molecular determinants––perhaps in a process similar to receptor “capping” on leukocytes––and can actually induce intracellular delivery. The subsequent intracellular itinerary of drug trafficking and its fate depend on the route of endocytosis and can be modulated by auxiliary pharmacological agents.
Vascular Targeting Of Anti-thrombotic Fusion Proteins
The use of whole IgG antibody molecules for EC targeting has some potential deficiencies, including Fc-fragment–mediated side effects. Production of antibody conjugates using chemical crosslinkers provides relatively heterogeneous formulations. In addition, rapid internalization of multivalent antibody conjugates, via mechanisms described above, makes them unsuitable for the delivery of drugs that supposed to act in the vascular lumen.
In this respect, an attractive alternative is provided by genetically engineered antibody fragments [such as scFv (consisting of both a VH domain and a VL domain)] that retain the specific antigen-binding affinity of the parent IgG (Figure 4A⇓). Such recombinant scFv can be genetically fused with protein drugs to produce high yields of relatively small (50–70 kD) homogeneous bi-functional molecules with favorable features including: 1) lack of Fc-fragment–mediated side effects; 2) relatively safe renal clearance; and 3) prolonged residence on EC surface, because monovalent scFv targeted to CAM does not induce CAM-dependent endocytosis. These features are ideal for endothelial targeting of anti-thrombotic drugs, such as fibrinolytics.

Vascular immunotargeting of recombinant fusion protein combining a single chain variable fragment of anti-PECAM and urokinase (scFv/uPA). A. A schematic representation of different antibody formats. Left side of panel shows generic structure of a whole immunoglobulin G (IgG) molecule, comprising two heavy and light chains linked by disulfide bonds. Hypervariable regions (CDRs) forming antigen binding sites are indicated. Right side of panel shows structure of a single-chain Fv (scFv) in which variable domains of light chain and heavy chain are covalently linked by a flexible inter-chain linker. As depicted, the size of scFv (30 kD) is six-fold smaller than whole IgG molecule (180 kD). B. A schematic diagram describing the cloning strategy for the fusion construct scFv/uPA. Variable domains of antibody heavy chain and light chain were linked and then fused to the N terminus of lmw-scuPA by a (Ser4Gly)2Ala3 linker. C. Pulmonary thrombolysis in mice by PECAM-specific scFv-uPA. The graph shows the dose-response curve of dissolution of pulmonary thrombi by bolus injection of equal doses of PECAM-specific scFv/uPA or non-targeted uPA in mice. Thrombolytic potency was expressed as percent of fibrinolysis vs dose administered. Dash line indicates spontaneous lysis. D. A simplified diagram of a proposed strategy for throm-boprophylaxis using vascular immunotargeting of genetically engineered scFv/uPA. scFv/uPA circulates in a form of a prodrug, binds to PECAM-1, and remains anchored on the luminal surface of endothelium for at least several hours. In situ thrombosis or embolism induces initial local conversion of plasminogen (Pg) into plasmin (Pn) by endogenous plasminogen activators (Endo-PA). Plasmin (and perhaps other enzymes) formed in the vicinity of the clot converts the endothelium-bound scFv/uPA into enzymatically active tcuPA, which in turn amplifies local formation of plasmin, reinforcing local thrombolysis, and preventing clot extension and reocclusion.
Thrombotic vascular occlusion by clots formed inside blood vessels is the leading cause of morbidity and mortality in cardiovascular pathological conditions and stroke. Fibrinolytic plasminogen activators (PA, serine proteases consisting of regulatory and catalytic domains) produce plasmin that subsequently degrades fibrin clots, leading to restored perfusion (88, 89). Plasminogen activators utilized for thrombolysis include tissue-type plasminogen activator (tPA) and single-chain urokinase type plasminogen activator (scuPA). The clinical utility of these drugs, however, is limited by 1) inadequate delivery owing to rapid elimination en route to the target and ineffective penetration into formed clots; and 2) side effects caused by collateral damage in the central nervous system and hemorrhages (88, 90, 91).
New DDS are being designed to achieve more effective and safe thrombolysis. Several groups have conjugated PA with antibodies directed to thrombi (e.g., activated platelets and fibrin) using chemical crosslinkers and recombinant fusion (92–94). For example, a series of recombinant fusion proteins were designed using fibrin antibody with tPA and or low molecular weight single-chain pro-uro-kinase plasminogen activator (lmw-scuPA) (92, 95). Construction of fusion PAs based on humanized fibrin antibody was also designed. In general, these fusion proteins exhibited catalytic activity and fibrin binding comparable to the parental PA and antibody and displayed higher potency as compared to the use of non-targeted PA in dissolving human plasma clots in vitro and in the rabbit jugular vein thrombosis model (93, 94).
The higher affinity of a fibrinolytic enzyme to clot components, however, the worse is its penetration of clots; hence, the targeting advantage is negated by an inadequate final step of delivery, which compromises the therapeutic effect. In principle, prophylactic administration of targeted thrombolytic agents could solve the problem of thrombus penetration. Pathological conditions and clinical settings characterized by a high propensity for recurrent thrombosis, in which such thromboprophylactic interventions would be most helpful, have been identified. However, no existing formulation prevents extravasation of PA into tissues (where PA induces side effects) or prolongs PA circulation sufficiently to permit their prophylactic utility. The conundrum could be solved by targeting PA to stably expressed endothelial determinants that are not internalized, boosting fibrinolytic activity in the vascular lumen and permitting prophy-lactic thrombolysis.
Only non-internalized determinants, such as PECAM-1, could be used for such targeting. Targeting anti-thrombotic agents to determinants that do become internalized (e.g., selectins) would lead, eventually, to their disappearance from EC surface and a loss of activity in the lumen (26). A recently designed PECAM-specific scuPA fusion protein seems to be well suited to achieve targeting to and prolonged binding to the EC surface (96). The variable domains of heavy chain and light chain (cloned from PECAM-specific anti-body producing hybridomas) that are connected with a [Gly4Ser]3 linker make up the PECAM-specific scFv, fused at its C-terminus with lmw-scuPA. The resultant homogeneous PECAM-specific scFv-uPA (scFv/uPA) (Figure 4B⇑) specifically binds to PECAM-expressing EC and retains fibrinolytic activity.
After intravenous injection, the fusion protein binds to endothelium and accumulates in the lungs (the preferential target of vascular immunotargeting to EC) of wild type, but not PECAM-null mice (96). The kinetic studies showed that scFv/uPA persisted in lung tissues for at least three hours and was retained on the surface of EC. In a mouse model of pulmonary thrombosis, scFv/uPA dissolved pulmonary emboli more effectively than did non-targeted uPA (Figure 4C⇑). Taken together, these data support the concept that anti-thrombotic therapeutics can be delivered in effective concentrations and for a relevant duration to the endothelial surface (Figure 4D⇑). Conceivably, scFv/uPA targeting to vascular EC may afford thromboprophylaxis in patients with acute risk of developing new or recurrent thrombi. It is conceivable that monovalent scFv-based DDS that target non-internalizable EC determinants (e.g., PECAM) can be utilized for delivery to the EC surface of anticoagulants (e.g., activated Protein C) or enzymes that decompose circulating harmful vasoactive peptides and toxic compounds.
Conclusion
Rational design of DDS targeted to any given cell type is a challenging endeavor that combines research efforts of experts in diverse areas including bioengineering, bioconjugation, nanotechnology, biomaterials, cellular recognition, and traffic (Figure 1⇑). Specific characteristics of a pathological process and cell type(s) that are the subject of a therapeutic intervention guide the path from target selection (Figure 1, I⇑) to fine-tuning of DDS formulation (Figure 1, IV⇑). Historically, concepts of drug delivery and targeting, and the ideology and methodology of DDS were initially conceived for the eradication of tumors or other pathogenic cells. A more recent paradigm of DDS targeting endothelial cells differs from these predecessors in several important ways.
In contrast with tumor cells poorly accessible to targeting, EC represent one of the best targets in the body: they are accessible to blood, represent a large surface area, and are amenable to the identification of surface determinants using high-throughput methods such as in vivo phage display. Size of DDS for EC targeting is limited only by circulation parameters (e.g., ~ 1 μm in diameter), which enhances the flexibility of selection and modulation of DDS parameters such as the subcellular localization of delivered drugs. Drug targeting to and across EC represents a globally important biomedical goal, owing to the key role of endothelium as an interface between blood and tissues.
On the other hand, safety of DDS targeting EC must be tested even more rigorously than that of DDS targeting tumors. Secondary toxic effects to tumor cells that arise from local or systemic activation of host defenses represent an unintended but welcome bonus. In most pathological conditions in which endothelial targeting would be helpful, such side effects would be detrimental. Thus, specific attention to these and other potential side effects, including effects of EC determinant functions inhibition or DDS remnants residing in the EC on their viability and functions is warranted.
Research of DDS targeting of the endothelium (e.g., vascular immunotargeting) currently enjoys an exponential rate of progress. Main areas of focused research include the new and better understanding of features of known endothelial target determinants, the molecular and nanoscale design of EC-targeted DDS, and the elucidation of mechanisms that control subcellular localization of drugs. For example, intracellular delivery of antioxidant enzymes using CAM-mediated endocytosis and surface anchoring of scFv-urokinase targeted to the same EC determinant illustrate the great biomedical potential of rationally designed DDS that target endothelial cells.
Acknowledgments
This work was supported by Pennsylvania NTI core project (TD and VM), AHA Scientist Development Grant 0435481N (SM) and grants from NHLBI RO1 HL71175, HL078785 and HL73940 and Department of Defense Grant PR 012262 (VM).
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Vladimir Muzykantov, MD, PhD, is an Associate Professor of Pharmacology and Medicine, a Senior Investigator in the Institute for Environmental Medicine, Director of Targeted Therapeutics Program in Institute for Translational Medicine and Therapeutics at the University of Pennsylvania (PENN) School of Medicine, in Philadelphia. He received his MD in Internal Medicine from the First Medical School in 1980 and PhD in Biochemistry from National Cardiology Research Center (both in Moscow, Russia). He held training and research positions at this center from 1980 (Senior Investigator from 1990) till 1993, when he moved to PENN. His research areas include targeted drug delivery and vascular biology. E-mail: muzykant{at}mail.med.upenn.edu; fax: 215 898-0868.

Silvia Muro, PhD, is a Research Assistant Professor of Pharmacology at the University of Pennsylvania (PENN) School of Medicine, in Philadelphia. She received her PhD from the Severo Ochoa Center for Molecular Biology in the Autonomous University of Madrid, Spain, in 1999, where she was a postdoc until 2001. She was a Postdoctoral Fellow from 2001 until 2003 and a Research Associate from 2003 until 2005 in the Institute for Environmental Medicine at PENN. She is working on mechanisms of endothelial targeting and metabolism of drug delivery systems, targeting of antioxidant therapies, and enzyme replacement strategies for lysosomal storage disorders. E-mail: silvia{at}mail.med.upenn.edu; fax: 215 898-0868.

Vladimir V. Shuvaev, MD, PhD, is a Research Investigator in Institute for Environmental Medicine at the University of Pennsylvania (PENN) School of Medicine, in Philadelphia. He received his MD in Biochemistry from Russian State Medical School in 1984 and PhD in Biochemistry from National Cardiology Research Center in 1992 (both in Moscow, Russia). He completed his post-doctoral training in University of Nancy (France) and Osaka University Medical School (Japan). In 2002, he joined PENN, where he is working on targeted delivery of antioxidant enzymes. E-mail: shuvaevv{at}mail.med.upenn.edu; fax: 215 898-0868.

Thomas D. Dziubla, PhD, is a Research Associate in the Institute for Translational Medicine and Therapeutics at the University of Pennsylvania (PENN) School of Medicine, in Philadelphia. In 2002, he received his PhD in Chemical Engineering from Drexel University, where he worked on porous soft coatings materials for implantable drug delivery devices. In 2002–2004, he was an NRSA postdoctoral fellow in the Institute for Environmental Medicine at PENN, where he worked on the design of degradable polymeric nanocarriers for the delivery of antioxidants. E-mail: dziubla{at}mail.med.upenn.edu, fax: 215 898-0868.

Bi-Sen Ding, BS, is a graduate student in the Graduate Group in Pharmacological Sciences, University of Pennsylvania School of Medicine, in Philadelphia. In 2000, he received his BS in Chemistry from Nanjing University, China, and in 2003, he received an MS in Molecular Medicine from the same university. Since 2004, he has been working on his PhD thesis (in the Muzykantov lab) on the design of therapeutic recombinant protein constructs targeted to endothelium. E-mail: ding2{at}mail.med.upenn.edu, fax: 215 898-0868.

