Iron Dysregulation and Neurodegeneration: The Molecular Connection
Abstract
Iron is essential for many biological processes however excess concentrations can be harmful to many tissues. Its amounts must therefore be carefully regulated in all cells of the body including those in the brain. Increased amounts of iron have been reported in many neurodegenerative disorders. Whether this increased iron contributes to neurodegeneration has been considered controversial. In this review, we discuss some recently identified anomalies in proteins linked with iron metabolism which signify a critical role for iron dysregulation in neurodegeneration.

Introduction
The management of iron is a critical issue for all living cells, where its presence is necessary for carrying out numerous vital biological reactions, but because of its highly reactive nature, iron is extremely toxic if its concentrations are not tightly regulated intracellularly. The divalent state of iron makes it highly toxic, as it can rapidly react with hydrogen peroxide and molecular oxygen to produce free radicals. Free radical formation can promote lipid peroxidation, DNA strand breaks, and modification or degradation of biomolecules, eventually leading to cell death. Cells have therefore evolved a sophisticated mechanism consisting of an array of proteins that regulate the amount of free iron in cells by modulating its uptake, storage, and export. Together, these proteins provide a state of cellular equilibrium whereby iron is readily available for all metabolic functions but sheltered from taking part in cytotoxic reactive oxygen species (ROS)-generating reactions (1, 2).
Iron plays crucial role in the brain where it is required to sustain the brain’s high respiratory activity, for myelinogenesis, and for the production of several neurotransmitters such as dopamine, norepinephrine, serotonin, and generation of γ-aminobutyric acid (GABA)ergic activity (3). In spite of its requirement, high levels of iron are of particular concern for the brain because of iron’s intense oxidative metabolism, which generates large amounts of ROS. In addition, the toxic effects of iron on brain cells are more pronounced because of their limited regenerative capabilities. It is therefore fortunate that iron must cross an additional obstacle in the form of the blood-brain-barrier to enter brain cells. The exact mechanism of iron uptake and export from the brain is not fully understood. Most of the proteins involved in the maintenance of iron metabolism are expressed in the brain, suggesting that brain cells follow similar homeostatic mechanisms as do all other cells in the body (4). It is also known that the brain accumulates iron in normal aging, which suggests that there is more import of iron into the brain than export. These increased levels of iron could place the brain iron homeostatic mechanism under pressure and make it more likely to err. Abnormal amounts of iron in the brain have been demonstrated in a number of age-related neurodegenerative disorders including Alzheimer Disease (AD) (5) and Parkinson Disease (PD) (6). Although it is generally acknowledged that this excess iron catalyzes the formation of ROS and induces oxidative damage to which the brain is particularly sensitive, brain iron accumulation itself has not, until recently, been widely considered a primary cause of neurodegeneration.
Proteins Involved In Iron Metabolism
The recent identification of mutations in genes encoding proteins involved in iron metabolism has supported a role for iron accumulation in neuronal death. Indeed, studies on these mutations (and other mutated genes whose actions appear to be more indirect) provide compelling evidence that dysregulation of iron can be a principal cause of neurodegeneration (3, 4, 7). In this review, we discuss some of the well-known molecules associated with iron metabolism in light of newly discovered mutations that have brought about a new appreciation of the connection between iron dysregulation and neurodegeneration.
Iron Regulatory Proteins
The iron regulatory proteins (IRPs) sense cellular iron concentrations and participate in the maintenance of cellular iron homeostasis. These cytoplasmic proteins bind to iron regulatory elements (IREs) present in mRNAs that encode proteins involved in iron metabolism thereby controlling their translation. Transferrin receptor (TfR) and ferritin are two important targets of IRPs and are largely responsible for cellular iron uptake and storage, respectively. The two forms of IRPs, IRP1 and IRP2, are structurally and functionally similar but differ in the way they sense the amount of cellular iron. IRP1 switches between two functions: 1) its IRE binding ability, and 2) the enzymatic function of a cytoplasmic aconitase. This switch of function depends upon the availability of iron, which allows the protein to assemble a functional iron-sulfur cluster essential for its aconitase activity or, in response to iron deficiency, to bind to IRE elements within the mRNAs of TfR and ferritin. IRP2, on the other hand, does not have an iron-sulfur cluster or aconitase function, but when iron is abundant, IRP2 is targeted for degradation by the 26S proteasome (8).
Genetic ablation of IRP1 in mice generates no overt pathology or neurological phenotype (9). IRP2 knock-out mice, however, develop a progressive neurodegenerative disease associated with dysregulation of iron metabolism in specific areas of the brain and the small intestine (10). IRP2−/− mice displayed abnormal accumulation of ferritin colocalized with iron in populations of degenerating neurons. Iron release caused by lysosomal degradation of iron-loaded ferritin has been proposed to result in oxidative damage leading to cell death. These mice show movement disorders including ataxia, tremor, and bradykinesia. IRP2−/−/IRP1+/− mice develop much more severe neurodegeneration characterized by widespread axonopathy and vacuolization in several brain regions including the substantia nigra (SN). Axonal degeneration takes place in tracts that are laden with ferritin and ferric (Fe3+) iron. Ultimately, neuronal cell bodies degenerate, especially in the substantia nigra, and microglia are activated to remove remnants of degenerating neurons. These mice display severe gait and motor impairment (11). Neurodegeneration and motor abnormalities occur only after significant iron accumulation has already taken place, suggesting that perturbation of iron accumulation caused by IRP disruption is a primary cause and not a secondary consequence of neurodegeneration.
Ferritin
Ferritin, an iron storage protein, is composed of twenty-four subunits consisting of both ferritin heavy polypeptide (H ferritin) and ferritin light polypeptide (L ferritin). H ferritin contains the ferroxidase activity that is essential for incorporation of iron into the protein shell whereas the L ferritin facilitates iron-core formation. Although the two polypeptides are encoded by different genes, translation of both proteins is controlled by common IRPs that bind to IREs in the 5′-UTR of the mRNAs (12). The ferroxidase activity of H ferritin has an essential regulatory role in iron metabolism. Deletion of the H ferritin gene in mice results in embryonic lethality (13). In contrast, increased or decreased expression of L ferritin in cellular models appears to have no evident effect on iron homeostasis (14), suggesting a less crucial role for L ferritin. Recently, however, several reports on ferritin-related neurodegenerative diseases or hereditary ferritinopathies have identified alterations in L ferritin conformation as critical to the development of neurodegeneration. Two types of mutations have been identified in the L ferritin gene (FTL) that result in neurodegeneration. The first one involves a nucleotide insertion (460–461InsA) that modifies the last twenty-two amino acids of L ferritin, causing a dominant adult-onset basal ganglia disease (15). Brain histopathology demonstrates cyst formations and cavitation in the basal ganglia, numerous iron-positive inclusions, and axonal swelling associated with neurodegeneration and observable movement disorder. The second mutation is also insertional and adds two nucleotides (498–499InsTC), altering the last nine amino acids and resulting in a similar dominant movement disorder (16). Brains from these patients display a mild cerebral and cerebellar atrophy and cavitation of the putamen. Studies on both the L ferritin variants suggest that the mutated proteins coassemble with normal H and L ferritins to form abnormal ferritin shells. These shells are unstable and either undergo expedited degradation or abnormal aggregation (17). Regardless of their intracellular fate, they significantly affect cellular iron homeostasis, causing an increase in intracellular iron availability and oxidative damage. It is hypothesized that in neurons, which contain predominantly H ferritin, the addition of a few mutant L ferritins is enough to destabilize the ferritin shells and accelerates their degradation rate. In long-lived cells like neurons, as opposed to fast dividing cells, some of the abnormal ferritin shells might escape degradation and are caused to aggregate by structural modifications as a result of the mutations. The post-mitotic status of neurons might therefore be conducive to such aggregations. Presence of ferritin body aggregates have been detected in patients suffering from neuroferritinopathies. These bodies contain iron, indicating that aggregation occurs after ferritin assembly and is consistent with the hypothesized mode of action of the mutations (17).
Another set of mutations in the IRE of the L ferritin is associated with hereditary hyperferritinemia and cataract syndrome. These mutations reduce the affinity of IRPs to bind IREs, resulting in constitutively high expression of ferritin as found in serum and tissues. Patients harboring these mutations exhibit early-onset of cataracts but no movement disorders (18).
Ceruloplasmin
Ceruloplasmin (Cp) is a serum α2-glycoprotein that binds ~95% of copper found in human plasma. The secreted form of this protein is found mainly in liver and serum whereas the glycosylphosphatidylinositol (GPI)-anchored form of Cp is expressed in astrocytes of the central nervous system (CNS). The discovery of aceruloplasminemia, an adult-onset, autosomal-recessive neurodegenerative disorder, revealed the iron regulatory system as the main target for dysfunction. Patients with this disorder lack serum Cp, owing to genetic mutations that introduce early translational termination, and symptomatically display both dementia and dystonia accompanied by low serum iron and mild anemia as well as elevation in serum ferritin. These results suggest that various tissues may be undergoing iron overload (19).
Knock-out Cp mouse models confirm the importance of Cp in iron homeostasis, albeit with a milder neurodegenerative phenotype (20, 21). Normally, Cp is expressed in the epithelial cells of the choroid plexus and astrocytes in the retina and basal ganglia (22). In the CNS of humans with aceruloplasminemia, iron is accumulated mostly in the retina and basal ganglia, further indicating the importance of Cp in the promotion of this iron-storage disorder. The GPI-anchored form of Cp is colocalized and physically associated with IREG1, an iron exporter, on the cell surface of astrocytes. Additionally, iron efflux from astrocytes is severely compromised in Cp knock-out mice (23). The mechanism underlying iron accumulation in Cp knockouts probably stems from an uptake of ferrous (Fe2+) iron into the affected tissues. A lack of Cp ferroxidase activity prevents the conversion of Fe2+ to the less toxic Fe3+ isoform (which would then normally bind to TfR for entry into the pool of recyclable iron). The presence of ferrous iron would then result in the generation of free radicals and accumulation of oxidative damage in the retina and basal ganglia leading to the macular- and neurode-generation observed in aceruloplasminemic patients.
Divalent Metal Transporter-1
Cellular uptake of iron occurs via the receptor-mediated uptake of TfR-bound ferric iron, acidification of the bound iron complex in endosomes, and transport of free ferrous iron into the cytosol via the divalent metal transporter-1 (DMT-1), also known as divalent cation transporter-1 (DCT-1), or natural resistance associated macrophage protein 2 (Nramp2). Both TfR and DMT-1 contain iron-responsive elements (IREs) on the 3′ UTR end of their respective mRNAs, resulting in a stabilization of message and increased translation of TfR and DMT-1 when intracellular iron levels are low. This regulatory mechanism ensures that a steady amount of iron gets imported into the cell for various cellular functions (24).
The importance of DMT-1/Nramp2 in iron transport is illustrated by the identification of a missense mutation of the Nramp2 gene in anemic mice that also exhibit defects in intestinal iron absorption and erythroid iron utilization (25). Impairments in cellular uptake of Tf-bound iron were observed in adult Nramp2−/− Belgrade rats, resulting in an iron-deficient state within the CNS as cells return the recycling endosome to the cell surface for recirculation (3, 26). Pharmacological blockade of DMT-1 could prove to be a therapeutic target for neurodegenerative disorders involving cellular iron overload; however, activation of the IRP system and increased expression of neuronal TfR can compensate for cellular iron depletion, resulting in an influx of iron. Sustained activation of IRP-1 and IRP-2 binding to DMT-1 mRNA is thought to underlie the increased amounts of DMT-1 protein demonstrated in rat hippocampus after excitotoxic kainate exposure (27). Increased DMT-1 would lead to elevated iron uptake and deposition into the cytosol (via DMT-1) without a concurrent increase in iron storage proteins (such as ferritin) to sequester the excess iron. Despite the absence of a clear neurodegenerative phenotype that results from a defect in DMT-1, it is clear that a balance in neuronal iron uptake and export is important in maintaining cellular functions while minimizing oxidative stress.
Frataxin
Friedreich’s ataxia is an autosomal recessive disorder that is characterized by neurodegeneration, cardiomyopathy, and diabetes. The primary causative factor in the onset of this disorder is a GAA nucleotide repeat expansion within the first intron of FRDA, resulting in the destabilization of frataxin mRNA and loss of protein expression (28). Frataxin mRNA is expressed predominantly in organs with high energy requirements and rich in mitochondria such as the brain, heart, liver, and kidneys. The presence of a mitochondrial targeting sequence in its N-terminal domain (29) and the accumulation of iron in yeast deficient in Yfh1p (the yeast homolog of frataxin) suggest that frataxin plays an important role in the homeostatic control of iron in mitochondria (30).
Frataxin is required for the biosynthesis of heme and iron-sulfur clusters that are necessary for the proper function of dozens of mitochondrial proteins. Interestingly, the crystal structure of frataxin resembles that of ferritin iron cores whereby monomeric frataxin acts to donate Fe(II) to ferrochelatase, and assembled frataxin can oxidize and store more than 2000 atoms of Fe(III) (31). Another essential function of frataxin is its ability to protect cells against oxidant-induced damage and promote cell life-span (32). Utilizing yeast mutational studies, Gakh et al. were able to differentiate between the ferroxidation and mineralization activities of frataxin (32). Impairments in mitochondrial bioenergetics resulting from iron-mediated oxidative damage remain a likely mechanism underlying neurodegenerative disorders such as FRDA and PD. What has been gleaned from studies on frataxin is that cells require this conserved protein for normal iron regulation and metabolism within their mitochondria.
Pantothenate Kinase 2
A group of disorders characterized by high iron concentrations in the basal ganglia, earlier known as Hallervorden-Spatz syndrome, has been recently renamed as neurodegeneration with brain iron accumulation (NBIA). The most prevalent form of NBIA is caused by mutations in a gene coding for pantothenate kinase 2 (33) and referred to as pantothenate kinase-associated neurodegeneration. The disease is inherited as an autosomal recessive trait which develops during the first two decades of life. The symptoms include progressive PD-like muscular rigidity, mental retardation, dystonia, and high concentrations of iron in the globus pallidus. In magnetic resonance imaging (MRI), these high levels of iron cause the medial globus pallidi to appear as bilateral hypointense (dark) regions with a central hyperintense (bright) region yielding a characteristic “eye of the tiger” image (34, 35).
Pantothenate kinase 2 (PANK2), is the first enzyme in the five step biosynthesis of coenzyme A, a key metabolic intermediate. Phosphopantothenate, the product of PANK2 activity, condenses with cysteine in the next step of coenzyme A synthesis (36) . PANK2, one of four human patothenate kinases, is the only isoform that localizes in mitochondria (37) and is the only isoform specifically expressed in the brain (38), which may account for the brain-specific pathophysiology of the disease. When PANK2 is mutated, phosphopantothenate is not produced; hence, cysteine tends to accumulate in the brain. The well known iron-chelating properties of cysteine could account for the observed iron overload: Cysteine undergoes rapid auto-oxidation in the presence of iron and induces free radical reactions. Within the brain, nonheme iron accumulates regionally and is the highest in the medial globus pallidus and the SN. Also, high concentrations of cysteine have been shown in globus pallidus (39), which could act to enhance iron-induced lipid peroxidation, further stressing a cell that has defective membrane biosynthesis arising from diminished coenzyme A synthesis. Although PANK2 is not directly involved in iron metabolism, it secondarily leads to: 1) iron deposition in the basal ganglia, 2) generation of reactive oxygen species (ROS), and 3) lipid peroxidation akin to aceruloplasminemia, a disease caused by a direct defect in iron homeostasis (35). Thus, iron dysregulation very likely participates in the pathogenesis of pantothenate kinase–associated neurodegeneration.
Hereditary Hemochromatosis (HFE) Protein
Hemochromatosis is an autosomal recessive hereditary disorder of iron metabolism wherein the body accumulates excess iron. Most cases are accounted for by two missense mutations (C282Y and C187G) in Hfe located on chromosome 6p (40). Mutations in the Hfe gene are the most common of all human genetic mutations, and estimates are that 20–40% of individuals of European ancestry carry at least one mutated Hfe allele (41–43). The normal HFE protein is thought to interact with the TfR, attenuating its capacity to mediate intracellular iron delivery. Excessive tissue iron sequestration may result from an abnormal interaction of the TfR with mutant HFE protein (40). Whereas iron overload in hemachromatosis is mostly reported in liver, heart, and pitutary glands, there have been both histochemical and MRI-based observations that also suggest iron accumulates in brains (44, 45). The presence of HFE protein has also been demonstrated on the surface of blood vessels, choroid plexus, and ependymal cells where it could influence brain iron uptake (46).
Its role in iron metabolism makes HFE a potential candidate gene in PD. Additionally, in hemochromatosis, clinical parkinsonism and iron deposits in the basal ganglia have been observed (47). Results of studies on the role of Hfe mutations in PD vary from a protective effect of C282Y heterozygosity (48) to no effect of either mutation (49). Alternatively, the C282Y mutation might increase the risk of developing PD or some form of parkinsonism (50).
The HFE protein has been detected by immunostaining in cerebral endothelium (51), a site known to contribute to neuronal injury in AD (52). There have been two reports suggesting that the presence of an Hfe mutation can influence the onset and incidence of AD (53, 54). Another study seeking linkage between AD and Hfe suggested a role for disrupted iron metabolism as a primary risk factor for neurodegeneration and oxidative stress in AD (55).
Heme Oxygenase-1
Heme oxygenase-1 (HO-1) is the rate-limiting microsomal enzyme involved in heme breakdown. It is also an important stress response protein that becomes activated in different cell types by a variety of oxidative insults. The by-products of the HO-1–mediated reaction (i.e., carbon monoxide, biliverdin, and ferrous iron) give insight into the potential regulatory functions of this protein. Biliverdin and its metabolite bilirubin have been thought to contribute to the protective antioxidant effects observed in in vitro and in vivo models; however, elevated HO-1 expression (and therefore activity) can also result in a pro-oxidant environment as a consequence of iron liberation (56).
Depending on the extent and duration of activation, fate of the by-products, and the cellular microenvironment, HO-1 may exert neuroprotective or detrimental effects. For example, targeted deletion of HO-1 in mice exacerbates cell death associated with serum deprivation, and treatment with iron chelators was able to block the iron accumulation and cell death associated with the HO-1 deficiency. Additionally, HO-1 overexpression augmented iron efflux from fibroblasts, further suggesting that the cytoprotective effects of HO-1 arises from a modulation of intracellular iron (57). On the other hand, mitochondrial ferrous iron accumulation appears highly dependent upon the increased expression of HO-1 in dopamine-exposed astroglia (58). The increase in mitochondrial ferrous iron could result in exacerbation of free radical formation and bioenergetic failure (i.e., the inability of mitochondrial capacity to generate ATP) as well as catalysis of catecholamine oxidation, leading to the formation of toxic quinones and other highly reactive species that amplify cellular oxidative damage.
Normally, HO-1 protein expression is restricted to a small subset of neurons and glia. Compared to age-matched normal controls, a significant elevation of HO-1 immunoreactivity was observed in astrocytes of AD hippocampi. Increased HO-1 expression was also demonstrated in the intracytoplasmic Lewy bodies found in PD brains (59). These affected brain regions are also observed to contain accumulated iron, thereby implicating HO-1 as a potential contributor to iron deposition in the CNS.
Metallothioneins
There are four isoforms of metallothioneins (MTs), small molecular weight and cysteine-rich proteins that bind metals such as zinc and iron in different tissues. MT-I and MT-II are expressed ubiquitously in all mammalian tissues, MT-IV in epithelial cells, and MT-III is predominantly found in the brain. Some of the main functions of MTs are to bind metal ions, regulate the biosynthesis and activity of metalloproteins, and to protect cells from oxidative damage by scavenging free radicals and unsequestered reactive heavy metals (60).
MT-III is also known as a growth inhibitory factor. It is abundant in normal tissue but is greatly reduced in astrocytes from AD brains (61), indicating that a disruption in metal homeostasis and redox status can result in neurodegeneration. The lipopolysaccharide (LPS)-dependent induction of MT-III in neuronal and astroglial cells in rats is compromised with aging (62). In animal models of PD, transgenic mice overexpressing MT1 were relatively resistant to the neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), whereas the double MT gene knockout (MT1 and MT2) mice exhibited augmented sensitivity to MPTP (63). Additionally, in vitro studies further implicated the neuroprotective role of MTs by demonstrating the attenuation of 3,4-dihydroxyphenylacetaldehyde (DOPAL, an endogenous toxic monoamine oxidase metabolite of dopamine)-induced apoptosis in rho− neurons (those lacking a mitochondrial genome) treated with MT oligonucleotides [as reported in (63)]. Although a direct link between MT and iron accumulation has not been established, it can be speculated that age-related decreases in MT induction contribute to alterations in homeostatic controls governing intracellular iron levels.
Conclusion
There has recently been a virtual surge in the discovery of molecules involved in iron metabolism. For a long time, ferritin, transferrin and transferrin receptor––the so called “traditional” molecules of iron metabolism––held court. The elucidation of their sophisticated regulation by IRPs, under normal and stressful conditions, established that the control of intracellular iron concentrations is vital for cell survival. With the discovery of other proteins (such as DMT1, ceruloplasmin, ferroportin, hephaestin, etc.), a more detailed picture of iron maintenance is beginning to be better appreciated, including the realization that there are likely more molecules involved in iron regulation that have yet to be identified . This is especially true for brain iron metabolism, which requires additional mechanisms that regulate the connection between systemic iron circulation and passage through the blood-brain-barrier to sustain the brain’s need for iron.
The identification of the mutations described above establish that mutations that specifically effect iron metabolism have the ability to affect the brain and cause neurodegeneration. This conclusion implies that iron overload in the brain––seen in many neurodegenerative disorders––could be causative and not just an inconsequential outcome. More research is needed to fully understand brain iron metabolism and the molecular mechanisms that cause specific brain areas to accumulate iron in different diseases. Of equal importance is the need to develop appropriate iron chelators whose effects, when properly titrated, effectively reduce iron concentrations in the brain but do not shift the brain towards undesirable iron deficiency. Currently, the metal chelator, clioquinol (64) and VK28 series of iron chelators (65) are being investigated for AD and PD, respectively.
Acknowledgments
This work was funded by the National Institutes of Health grant NS41264 (JKA).
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Deepinder Kaur, PhD, is a staff research investigator in the lab of Julie Andersen at Buck Institute for Age Research. She received her PhD from Jawaharal Nehru University, New Delhi, India where she trained in developmental and molecular biology. Her current research interest is to understand the basis of iron dysregulation in Parkinson Disease and probe its contribution to development of the disease.

Julie Andersen, PhD, is a Professor at the Buck Institute for Research in Aging in Novato, California. She received her PhD from the Department of Biological Chemistry in the UCLA School of Medicine. She was a postdoctoral fellow in the laboratory of Dr. Xandra Breakefield at Massachusetts General Hospital as part of the Harvard Neurology Program from 1989 until 1993 when she joined the Department of Gerontology at the University of Southern California as an Assistant Professor. She was promoted to Associate Professor in 1999 and joined the faculty of the Buck Institute in 2000 where she was promoted to Full Professor in 2000. Dr. Andersen’s research interests encompass the molecular and cellular mechanisms that give rise to Parkinson Disease. E-mail: jandersen{at}buckinstitute.org; fax: 415-209-2231.

Donna W. Lee, PhD, is a postdoctoral fellow in the laboratory of Dr. Julie K. Andersen at the Buck Institute for Age Research. Formally trained as a neurotoxicologist at the University of Rochester, NY, her research interests focus on the contribution of environmental factors on the dysregulation of oxidative stress and iron regulatory pathways in neurodegenerative models. Her other passions include the Brazilian martial art Capoeira (performed to music) and cooking.
Proteins Involved in Iron Regulation
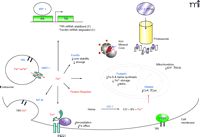
Intracellular fate of ferrous iron. The uptake of iron in the ferric form (Fe3+) is mediated by the transferrin receptor (TfR) and the reductase present in acidified endosomes converts Fe3+ to the ferrous form (Fe2+) before transport into the cytosolic space by divalent metal transporter-1 (DMT-1). The resultant Fe2+ within the labile iron pool (LIP) either: 1) activates the regulatory iron regulatory protein/iron regulatory element (IRP/IRE) system, 2) gets sequestered by chaperones or storage proteins [such as ferritin, metallothioneins (MTs), and frataxin], 3) reoxidizes into Fe3+ and effluxes out of the cell via Cp and IREG1, or 4) participates in iron-catalyzed reactions to generate reactive species [e.g., pantothenate kinase 2 (PANK2)-deficient oxidation of cysteine residues]. Additionally, Fe2+ can be liberated from free heme groups to contribute to the LIP. Heph; hephaestin, a membrane-bound protein homolog of Cp.

