NEUROGENIC INFLAMMATION AND MIGRAINE: IMPLICATIONS FOR THE THERAPEUTICS
Abstract
Significant recent advances in molecular pharmacology have elucidated the molecular pathways involved in neurogenic inflammation (NI). The release of tachykinins and endothelin-3 (ET-3) from trigeminal neurons induces dural vascular permeability and vasodilatation via activation of tachykinin receptor 1 (Tacr1) and endothelin receptor type B (Ednrb) on endothelial cells. Endothelial cell receptor stimulation results in cellular contraction, leading to plasma protein extravasation (PPE), which is the most recognized physiological hallmark of NI, and nitric oxide–induced vasodilatation. By contrast, the release of calcitonin gene–related peptide (CGRP) from trigeminal neurons—also a key physiological component of NI—does not affect vascular permeability but does induce neurogenic vasodilatation (NV) via the direct, (i.e., endothelium-independent) relaxation of vascular smooth muscle. The molecular pharmacology of NI is discussed within the context of migraine research and assesses the putative role of the two key physiological components of NI (i.e., PPE and NV) in migraine pathophysiology. The data indicate that the PPE component of NI plays no significant role in migraine but that NV is likely to be involved in migraine pathophysiology.

Introduction
Migraine is a clinical syndrome of self-limited neurogenic inflammation (1).
In 1910, Bruce observed that the application of mustard oil to the conjunctival sac in experimental animal models produced inflammation that could be blocked by sensory nerve ablation (2, 3). These early studies of the sensory neuron–induced “wheal and flare” reaction led to the concept of neurogenic inflammation (NI), referring to both vasodilatation and increased vascular permeability arising from the “retrograde” release of neuropeptides by capsaicin-sensitive sensory neurons in the periphery (4). NI was thus recognized as a physiological process of inflammation induced by the nervous system.
The modulation of both local blood flow and vascular permeability by small-fiber sensory neurons (C fibers and some Aδ fibers) is now well-established. At the molecular level, this modulation is mediated by the peripheral release of neuropeptides such as substance P (SP), neurokinin A (NKA), endothelin-3 (ET-3), and calcitonin gene–related peptide (CGRP). Direct chemical, thermal, or electrical stimulation of sensory nerves in rodents induces vascular permeability changes that are mediated predominantly by the tachykinins (e.g., SP and NKA) and vasodilatation (i.e., the axon reflex flare) that is mediated predominantly by CGRP (5).
A possible relationship between NI and migraine was first suggested by Dalessio (1, 6), who stated that migraine occurs as a result of vasodilatation associated with a sterile local inflammatory reaction. In 1984, Moskowitz extended this relationship when he proposed a mechanism for migraine headaches that involves depolarization-induced release of SP or other neuropeptides from trigeminal terminals; these neuropeptides were postulated to increase vascular permeability and dilate cerebral blood vessels (7). The NI theory of migraine was advanced considerably by the development of rodent models of NI that allowed for the detection of plasma protein extravasation (PPE) in the dura following chemical, electrical, or immunological stimulation (8–17). Vascular permeability changes consistent with “inflammation” (i.e., PPE) and induced by SP released from trigeminal neurons was posited to play a key role in migraine pathophysiology with either a limited or negligible role for neurogenic vasodilatation (NV) induced by CGRP (18).
Despite the continued widespread acceptance of NI as an essential component of the pathophysiology of migraine (19–25), a definitive role for dural NI in the pathophysiology of migraine remains unresolved. Support for the hypothesis has been based, to a significant degree, on the fact that some effective antimigraine agents (e.g., ergots and triptans) inhibit dural NI in animals following trigeminal stimulation (18). In the past decade, however, various arguments against the involvement of NI in migraine have been developed (26–32). These arguments are based, to a large degree, on the fact that numerous drugs that block the PPE component of NI in animals have failed to demonstrate antimigraine efficacy in human clinical trials.
An underappreciated fact is that NI consists of two distinct and independent physiological components (i.e., PPE and NV) (Figure 1⇓). Drugs that inhibit the release of multiple trigeminal neuropeptides (e.g., SP, NKA, ET3, and CGRP) block both the tachykinin-induced PPE and CGRP-induced NV components of NI. Thus, these drugs (e.g., triptans, ergots) are unable to elucidate whether their clinical antimigraine efficacy might derive from the inhibition of both the PPE and NV components of NI, or from an inhibition of only one of the two components of NI (Figure 1⇓).
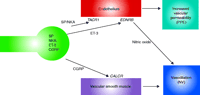
Molecular pathways of neurogenic inflammation. Neurogenic inflammation consists of two physiological arms, namely, plasma protein extravasation (PPE) and neurogenic vasodilatation (NV). The release of neuropeptides from trigeminal neurons (green) can be stimulated or inhibited by distinct agents. Both the endothelium and vascular smooth muscle can contribute, differentially, to neurogenic inflammation. The endothelium, for example, can promote vasodilatation through the use of NO as a messenger, whereas vascular smooth muscle may contribute directly to vasodilatation. (SP, substance P; NKA, neurokinin A; ET-3, endothelin-3; CGRP, calcitonin gene–related peptide; TACR1, tachykinin receptor 1; EDNRB, endothelin receptor type B; CALCR, calcitonin receptor.)
A more definitive resolution of the mechanisms underlying migraine has many practical and potentially important implications. An understanding of the roles of the two distinct physiological components of NI in migraine could allow for greater insights into the pathophysiology of migraine, and this information could be instructive in better exploiting existing therapeutic agents to combat migraine. An objective evaluation of NI in migraine should also provide an assessment of the ability to extrapolate data from various laboratory models of NI to the human condition. The design of novel strategies against migraine will also depend on a deeper understanding of pathophysiological mechanisms and their assessment in non-human models.
Substance P–Induced PPE in Neurogenic Inflammation
The key physiological hallmark of NI that can be measured quantitatively is PPE. Following stimulation of peripheral sensory nerves in rodents, PPE occurs in the postcapillary venules (33, 34). PPE is accompanied by increased blood flow, which results from dilatation of arterioles. In rodent skin, for example, NI is manifested as both a wheal (i.e., PPE) and a flare (i.e., vasodilatation). SP is the primary mediator responsible for PPE, whereas both CGRP and SP induce vasodilatation (5, 22, 35–37), albeit via distinct molecular mechanisms (Figure 1⇑). Specifically, the PPE component of NI is predominantly the result of tachykinin activation of Tacr1 on the endothelial cells of postcapillary venules and collecting vessels (38, 39). Within seconds of Tacr1 activation, endothelial gaps (ca 0.5 to 1.5 microns in diameter) form that allow for the extravasation of plasma proteins. The gaps that form in response to SP are fully reversible and are located at the intercellular junctions of endothelial cells (38, 39). SP-induced vasodilatation is endothelium-dependent and results from nitric oxide release (40).
Gene knockout studies have confirmed that the tachykinins are the primary mediators of the PPE component of NI. For example, disruption of the mouse preprotachykinin A (Tac1) gene, which can normally undergo alternative splicing to generate both SP and NKA, impairs neurogenic inflammatory responses but does not affect non-neurogenic inflammatory stimuli (41, 42). In Tac1-deficient mice, NI was reported to be minimal (41), such that PPE was profoundly reduced, relative to wild-type mice, following the topical application of capsaicin to the ear (41). By contrast, inflammation induced by complete Freund’s adjuvant, which results from mast cell degranulation and is therefore non-neurogenic, is unaltered in Tac1-deficient mice (41). Consistent with these observations is the fact that mice lacking the Tacr1 fail to develop PPE in response to capsaicin (43, 44).
Based on data with endothelin antagonists, it is likely that primary sensory neuron release of ET-3 accounts for the residual NI in Tac1-deficient animals (45). Nevertheless, gene knockout studies confirm that Tac1 expression accounts for the majority of NI in rodents via stimulation of Tacr1 on endothelial cells.
CGRP-induced NV in Neurogenic Inflammation
CGRP is a potent vasodilator that is released from sensory neurons, often concurrently with tachykinins and ET-3, and is the primary mediator of neurogenic vasodilatation (NV). Although NV can be induced by stimuli that induce NI in rodents (e.g., antidromic stimulation, capsaicin), it is mediated primarily by CGRP-induced relaxation of vascular smooth muscle (5, 46, 47). More specifically, CGRP induces NV via a direct stimulation of GPCR-coupled receptors (i.e., CALCR) linked to the stimulation of adenylate cyclase on vascular smooth muscle. Thus, the vasodilatory effect of CGRP is not dependent on endothelium in humans (Figure 1⇑). A recurring misconception in the literature is that CGRP induces NI (25). However, CGRP does not induce NI in humans or rodents, either directly (via effects on endothelial cells) or indirectly (via effects on mast cells). Indeed, even at micromolar concentrations, CGRP induces vasodilatation but not PPE in humans (48–52) and rodents (8, 53, 54).
Hypothesis Testing of the “Neurogenic Inflammation Theory of Migraine”: Selective Inhibition of Dural PPE
A putative role for NI in the pathophysiology of migraine was first proposed thirty-five years ago. The hypothesis evolved to state that migraine is characterized by the release of SP and other neuropeptides from trigeminal sensory neurons resulting in a secondary inflammation within the dura. This hypothesis gained widespread, but not complete, acceptance by the 1990s and was instrumental in guiding the development of potentially novel therapeutic approaches to migraine for more than a decade.
The most frequently studied approach to the pharmacological inhibition of dural NI has been the stimulation of inhibitory receptors located on presynaptic trigeminal neuronal terminals. However, these G protein–coupled receptors (e.g., 5-HTR1B, ADRA1A) inhibit the release of all sensory neuropeptides (Figure 1⇑). As a result, this pharmacological mechanism blocks both PPE (as a result of the inhibition of both tachykinin and ET-3 release) (55–58) as well as NV (as a result of the inhibition of CGRP release). Therefore, pharmacological agents that act via presynaptic inhibition of all sensory neuropeptide release inhibit both PPE and NV.
By contrast, pharmacological approaches have been identified that can differentially inhibit the PPE and NV components of NI. Such agents allow for an analysis of whether one or both of the physiological components of NI might be involved in migraine pathophysiology. For example, dural NI in rodents can be inhibited by three distinct pharmacological approaches that selectively inhibit PPE but do not affect CGRP-induced NV (Table 1⇓). Specifically, drugs that block TACR1 and EDNRB receptors on endothelial cells selectively prevent tachykinins and ET-3, respectively, from inducing endothelial cell–mediated PPE and vasodilatation but have no effect on the NV induced by CGRP release. A third pharmacological mechanism involves agents, such as CP-122,288 and 4991W93, that act on “extravasation receptor(s)” in trigeminal neurons to prevent PPE but not NV (59).
Three Mechanisms for the Selective Inhibition of Dural PPE
All three of these mechanistic approaches have now been investigated in human migraine studies. In total, eight pharmacological agents (described in detail below) have been developed that demonstrate the preclinical ability to selectively inhibit dural PPE but not trigeminal-induced NV. These agents are extremely relevant to the “NI Theory of Migraine” because their ability to inhibit PPE but not NV in animal models predicts that they would be effective anti-migraine agents in humans, if PPE played a role in migraine pathophysiology. Therefore, these drugs provide a set of powerful tools that have been used to test the “NI Theory of Migraine.”
Antagonists of TACR1
TACR1 antagonists are unequivocally effective in blocking dural PPE (Figure 1⇑) following electrical, chemical, or mechanical activation of the trigeminal system (60). Given the primary role of TACR1 receptors in the molecular cascade of NI, multiple pharmaceutical companies developed selective, potent and brain-penetrant TACR1 receptor antagonists in the 1990s and tested their clinical effects in migraine. Although it is often stated in the literature that these agents block NI, in reality their effect is limited largely to inhibition of the PPE component of NI. They have no effect on the CGRP-induced NV component of NI.
Several drugs were hypothesized to be highly effective in the acute treatment of migraine because they selectively inhibited trigeminal-induced PPE in rodent models. These potent and selective TACR1 antagonists include lanepitant (i.e., LY-303870) (61), GR205171 (62), L-758,298 (a prodrug that gives rise to the potent and brain-penetrant NK receptor antagonist L-754,030; also called MK0869) (63), FK 888 (64), and dapitant (i.e., RPR-100893) (60). The clinical studies are summarized below.
Lanepitant (i.e., LY-303870)
Lanepitant was evaluated in a randomized, double-blind, placebo-controlled crossover study to determine its effect in reducing migraine pain and associated symptoms (65). Outpatients (n = 40) treated four migraine headaches of moderate to severe pain with oral doses of the drug. Lanepitant had no statistically significant difference versus placebo in improving migraine pain. There was no change in migraine-associated symptoms, and no adverse events could be attributed to lanepitant. The authors concluded that lanepitant was not effective in the acute treatment of migraine (65). Moreover, lanepitant was also found to be ineffective in a migraine prevention study (66).
GR205171
A randomized, double-blind, placebo-controlled clinical study of the acute treatment for migraine involved the effect of a single intravenous dose of GR205171 (n = 31) or placebo (n = 32) (67). There was no significant difference in reduction of headache severity between GR205171 and placebo at any time point. The authors concluded that GR205171 was not effective in the acute treatment of migraine and questioned the importance of tachykinins, acting via peripheral or central TACR1 receptors, in the pathophysiology of migraine (67).
L-758,298
Intravenous L-758,298 was studied in a placebo-controlled study (n = 72) in the acute treatment of migraine (68). Subjects with moderate to severe migraine received an IV infusion (20–60 mg) of L-758,298 or placebo. Statistical evidence of efficacy was based on patients treated with L-758,298 (60 mg) or its corresponding placebo. Two hours after treatment, 13 of 39 patients (33%) on L-758,298 and 11 of 21 (52%) on placebo reported pain relief, a difference that was not statistically significant. The authors concluded that L-758,298 was not effective as an acute migraine treatment (68).
FK 888
Clinical migraine trials using FK 888, a TACR1 antagonist, have been conducted in Japan and Europe (69). Clinical efficacy was not observed in the Phase-II trials; therefore, clinical development of FK 888 for migraine was discontinued (69).
Dapitant (i.e., RPR-100893)
A double-blind, randomized, placebo-controlled, dose-finding, parallel-group study was conducted to compare three oral doses of dapitant and placebo in patients (n = 139) with moderate to severe migraine attacks (70). No effect on headache intensity was observed among the four treatment groups. The authors concluded that dapitant is not effective in the acute treatment of migraine (70).
Antagonists of EDNRB: Bosentan
The effects of EDNRB antagonists have been tested in rat models of NI and non-neurogenic plasma extravasation in the dura mater and extracranial tissues (45). Specifically, both bosentan (a nonselective endothelin receptor antagonist) and Ro 46-8443 (a selective EDNRB antagonist) inhibit dural PPE induced by unilateral electrical stimulation of the trigeminal ganglion (45). Bosentan is also effective in inhibiting dural PPE induced by intravenous injection of capsaicin, whereas it is ineffective in extracranial tissues or after the induction of non-neurogenic inflammation by SP. By contrast, a selective EDNRA antagonist, BQ-123, has no effect in this animal model of NI. These data indicate a specific role of EDNRB in mediating PPE following stimulation of the trigeminal ganglion (45).
Based directly on the similarity of the pharmacological activity of bosentan in animal models of NI to known antimigraine agents such as ergots and triptans (45), the effect of the antagonist was studied in the acute treatment of migraine in a randomized, double-blind, placebo-controlled, clinical trial (27). Clinical improvement was observed in 22% of the bosentan-treated and in 36% of the placebo-treated patients. This negative finding led the authors to conclude that inhibition of dural NI may not predict the clinical efficacy of experimental antimigraine drugs (27).
Prevention of NI-inducing Neuropeptide “Extravasation” in Trigeminal Terminals
A group of pharmacological agents have been identified that are extremely potent inhibitors of neurogenic PPE in animal models (71, 72). These agents include CP-122,288, CP-122,638, 4991W93, and 5-carboxamidotryptamine. The ability of CP-122,288 and 5-carboxamidotryptamine to inhibit dural NI, unlike sumatriptan, is not blocked by selective 5-HT 1B/1D antagonists (73), indicating that this pharmacological effect is not mediated by 5-HT1B or 5-HT1D receptors. It has been suggested that these agents act at “extravasation receptors” in trigeminal neurons (59). At the low doses needed to inhibit NI, they have no effect on neurogenic vasodilatation (59).
CP-122,288
The acute antimigraine efficacy of intravenous and oral CP-122,288 has been evaluated in two double-blind studies (30). In a crossover design, patients randomly received CP-122,288 intravenously, placebo, or both. In an oral study, subjects received placebo or one of four doses of CP-122,288. Both studies were stopped prematurely when target efficacy could not be achieved. The authors concluded that CP-122,288 was not clinically effective at doses and plasma concentrations in excess of those required to inhibit neurogenic PPE in animals (30).
4991W93
Results from a phase-II double-blind, placebo-controlled design study of intravenous 4991W93 in the acute treatment of migraine has been reported (74). There was no observed clinical benefit in migraine using two different doses of the drug.
Hypothesis Testing of the “Neurogenic Inflammation Theory of Migraine”: Selective Inhibition of CGRP-induced NV
A role for CGRP-induced NV in the pathophysiology of migraine is supported by the results of clinical studies using a selective CGRP receptor antagonist. Olcegepant (i.e., BIBN 4096 BS) was reported to be effective in the acute treatment of migraine for up to six hours after onset (75). In an international, multicenter, double-blind, randomized clinical trial of BIBN 4096 BS, 126 patients with migraine received either placebo or between 0.25 and 10mg intravenously over a period of ten minutes. A group-sequential adaptive treatment-assignment design was used to minimize the number of patients exposed. A dose of 2.5 mg evoked a significant response rate [66% vs 27% placebo (p = 0.001)]. The BIBN 4096 BS group as a whole had a response rate of 60 percent. Significant superiority over placebo was also observed with respect to most secondary end points: the pain-free rate at two hours; the rate of sustained response over a period of twenty-four hours; the rate of recurrence of headache; improvement in nausea, photophobia, phonophobia, and functional capacity; and the time to meaningful relief. A positive effect was apparent after thirty minutes and increased over the next few hours. The overall rate of adverse events was 25% after the 2.5-mg dose of the drug (mostly parasthesias) as compared with 12% for placebo. There were no serious adverse events. The authors concluded that the CGRP antagonist BIBN 4096 BS was effective in treating acute attacks of migraine (75).
Conclusion
The “NI Theory of Migraine” predicts that inhibitors of dural NI in animal models should be effective in the acute treatment of migraine. NI, however, consist of two major physiological components: PPE mediated by tachykinins and ET3 and NV mediated predominantly by CGRP effects on vascular smooth muscle. Eight drugs that are inhibitors of PPE, but not NV, in animal models have now been studied in clinical migraine trials. These eight drugs utilize at least three distinct molecular pharmacological mechanisms of action. None of these drugs have been reported to be effective in the acute treatment of migraine. By contrast, a single drug that inhibits CGRP-induced NV has been shown to be effective in the acute treatment of migraine (75). Therefore, based on these numerous human clinical studies, it can now be concluded that the ability of drugs to selectively inhibit dural PPE in rodents has no predictive value in the acute treatment of migraine. However, the NV component of NI remains a molecular pathway that may play a key role in migraine pathophysiology.
A variety of other data are inconsistent with a role for PPE in migraine. For example, if PPE (and specifically SP) were involved in the pathophysiology of migraine, then increases in tachykinin levels should be detectable during a migraine attack. Plasma levels of various neuropeptides in the extracerebral circulation of humans were determined in patients who had migraine (76). Venous blood was sampled from both the external jugular and cubital fossa ipsilateral to the side of headache. Plasma levels of SP, neuropeptide Y, vasoactive intestinal polypeptide, and CGRP were determined, and a substantial elevation of the CGRP level in the external jugular but not the cubital fossa blood was seen in both classic and common migraine. The levels of SP were unaltered (76), providing further evidence against a functional role for SP in the pathophysiology of migraine.
Striking species variations in the PPE component of NI are likely to underlie the failure to extrapolate animal model data to human migraine clinical efficacy. Indeed, the PPE component of NI may not even exist in humans except in rare and extreme situations. For example, capsaicin induces pain and vasodilatation in human skin but it does not induce PPE or histamine release from human mast cells (77–80). In human skin, physiological concentrations of either SP or CGRP (< 10−9 M) applied intradermally induce dose-dependent local vasodilatation but not PPE (51). By contrast, intradermally injected SP (10−13 M) in guinea pig skin induces PPE that can be blocked by SP antagonists (81). In human oral mucosa, activation of local primary sensory neurons induces local vasodilatation (82). Capsaicin (3% solution) stimulation of the human oral mucosa induces mild pain and a pronounced vasodilatation, but not edema, in the vicinity of the stimulus (82). By contrast, in the rat, topical application of capsaicin onto the oral mucosa (83) induces PPE as a result of tachykinin release from sensory neurons (84, 85). Thus, rodent endothelial receptors for the tachykinins appear to be significantly (i.e., orders of magnitude) more sensitive than human TACR1. The key assumption of the “NI Theory of Migraine” is that trigeminal terminal release of SP and possibly additional neuropeptides into the dura induces inflammatory changes that are hallmarked by PPE. Trigeminal sensory neuronal release of the tachykinins and ET3 in laboratory animals is the absolutely essential molecular event that initiates the physiological cascade of events that result in dural PPE as a result of endothelial cell stimulation. Data that support the existence of this physiological event in humans, as opposed to rodents, are simply lacking. The failure of the eight drugs described above to show any efficacy in migraine suggests that the PPE component of NI may be far more relevant to lower mammal physiology than to humans. Thus, the dural “NI Theory of Migraine,” specifically as it predicts the induction of dural PPE in humans during a migraine attack, is no longer tenable. The scientific insights gained from the animal studies of PPE have been significant and valuable, but their relevance to humans with migraine is limited. By contrast, the search for additional agents that can inhibit the CGRP-induced NV component of NI seems warranted.
Acknowledgments
The author has no financial support from, or equity position in, manufacturers of products mentioned in this manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Stephen Peroutka, MD, PhD, is Franchise Development Leader for Pain at Johnson & Johnson Pharmaceutical Research Development. He earned his doctoral degrees from the Johns Hopkins University School of Medicine, and he has served in directorial positions in both academia and industry. E-mail DrPeroutka{at}aol.com; fax 609-730-3538.

