The Cardiotoxicology of Anthracycline Chemotherapeutics: TRANSLATING MOLECULAR MECHANISM INTO PREVENTATIVE MEDICINE
Abstract
Anthracyclines remain a mainstay of chemotherapy in spite of their well-recognized cardiotoxicity. Recent experience with trastuzumab (Herceptin) and anthracycline therapy has prompted a detailed analysis of the function of erbB2 in the heart. These studies demonstrate a cardioprotective effect of neuregulin, the endogenous ligand for the erbB4/erbB2 heterodimeric receptor complex. Although the mechanisms of cytoprotection remain incompletely understood, these studies have triggered the question of whether physiological manipulation of cardioprotective pathways that involve erbB can be used to improve outcome in patients treated with anthracyclines. The local activation of cardioprotection by cardiovascular exercise may be such a manipulation and warrants further investigation.
Introduction
Anthracycline antibiotics such as doxorubicin (i.e., adriamycin) and daunorubicin are some of the most effective chemotherapeutic agents used in the treatment of cancer; however, the utility of these pharmaceuticals is limited by cumulative, dose-related, progressive myocardial damage that may lead to congestive heart failure (CHF). Thus, the oncologist prescribing anthracyclines must constantly weigh the beneficial (i.e., anticancer) effects of the drug against the risk of cardiac toxicity. Much has been learned about the mechanisms of anthracycline cardiotoxicity since its first recognition. In spite of this knowledge, an acceptable strategy for prevention of anthracycline-induced cardiac toxicity, other than limitation of anthracycline exposure, remains lacking. Physicians treating patients whose tumors are responding to anthracyclines are often faced with the difficult question as to whether the risk of heart failure outweighs the benefits of further cancer therapy. As the incidence of cancer rises, so too does the incidence of anthracyclineinduced heart failure, which currently represents ~1% of patients with advanced heart failure due to ventricular systolic dysfunction. Recently, a novel drug targeting the erbB2 oncogene has been shown to increase the risk of anthracycline cardiotoxicity. This observation, together with work in animal and cell models, sheds new insight on how anthracycline toxicity is modulated by endogenous cardiac growth and survival signaling pathways. These observations lead to the hypothesis that physiological activation of these endogenous factors may yield clinically useful strategies for cardioprotection against this predictable cardiac insult.
Overview of Anthracycline Cardiotoxicity
Anthracycline antibiotics represent a major class of antitumor drugs and have been used in chemotherapy for over thirty years. They are highly effective and widely used in the treatment of leukemias, lymphomas, and adenocarcinomas. The mechanisms of cytotoxicity of anthracyclines in cancer cells include: 1) intercalation in DNA, leading to inhibition of both DNA replication and RNA transcription; 2) generation of free radicals, leading to DNA damage or lipid peroxidation; 3) DNA binding and alkylation; 4) DNA cross-linking; 5) interference with DNA unwinding, DNA strand separation, and helicase activity; 6) direct membrane damage due to lipid oxidation; and 7) inhibition of topoisomerase II. In response to some or all of these effects, tumor cell growth is inhibited and cells are more likely to die by one or more mechanisms. [For review, see (1).]
A major limitation of anthracycline use is a cumulative dosedependent cardiac toxicity (2) (Figure 1⇓). Early strategies to prevent cardiac toxicity included reductions in single doses of anthracyclines, as well as prolonging the infusion of drug to limit peak serum concentrations. Despite these efforts, the cardiotoxicity remains. A large-scale study of 630 patients randomized to a doxorubicinplus- placebo arm of three phase III studies, over the years of 1988 to 1992, reported a 5.1% risk of congestive heart failure in patients receiving cumulative doses up to 400 mg/m2, rising to 48% in patients receiving doses up to 700 mg/m2. Congestive heart failure was reported in patients receiving as little as 300 mg/m2. Advanced age remains a significant risk factor, with patients older than sixtyfive years having triple the risk of doxorubicin-related CHF compared with younger patients at the same cumulative dose (3).
The cytotoxicity of anthracycline antibiotics occurs in normal tissues as well as the tumor target, and the effects on the heart pose a major clinical dilemma.
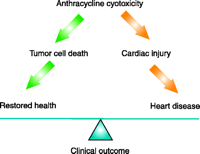
Anthracyclines have long been thought to induce cardiotoxicity by mechanisms other than those mediating their antitumor effectiveness, a concept that raises hope for the development of strategies that protect the heart without diminishing tumor response (2). Multiple proposed mechanisms of anthracycline-induced cardiac cellular injury have been based upon studies in animals and cell cultures, although it remains unclear which of these is at work in the clinical context of anthracycline use. Most mechanisms propose the induction of oxidative stress by the anthracyclines, but it is not clear why this would result in preferential cardiotoxicity. Anthracyclines induce membrane damage via lipid peroxidation in all tissues, including the heart (4, 5). Whereas formation of reactive oxygen species are induced by the quinone moiety of anthracyclines, oxidative stress can also occur via induction of nitric oxide synthase, leading to nitric oxide and peroxynitrite formation. This latter mechanism has been linked to nitration and inactivation of key enzymes in the heart including myofibrillar creatine kinase (6). Anthracyclines also cause impairment of membrane binding, assembly, and enzymatic activity of mitochondrial creatine kinase, although the consequences of this impairment are yet unclear (7). In the heart, like other tissue, anthracyclines intercalate into nucleic acids, causing suppression of DNA, RNA, and protein synthesis (8). Some transcriptional regulatory proteins that appear important for regulation of cardiac specific genes appear particularly susceptible to anthracyclines (9–11). This leads to impaired synthesis of myofilament proteins, which in the presence of accelerated myofilament degradation (12) creates a net negative balance of sarcomeric proteins, a “cardiac sarcopenia” (Figure 2⇓). Myocyte cell death by both apoptosis and necrosis has also been implicated, and the net loss of cells may contribute to “cardiac wasting.” Finally, anthracyclines induce changes in adrenergic function and adenylate cyclase (13–15) and abnormalities in Ca2+ handling (16), all critical for the dynamic regulation of cardiac function. The extent to which each of these contributes to dosedependent heart failure in anthracycline-treated patients remains controversial.
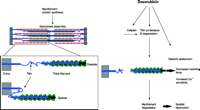
Mechanistic role of titin and anthracycline-induced cardiotoxicity. Myofilament damage is a key element in anthracycline-induced cardiotoxicity, which along with reduced myofilament protein synthesis contributes to the cardiac injury that begets heart failure. Specifically, treatment of myocytes with doxorubicin induces breakdown of the giant myofilament protein titin (185 kDa), ultimately leading to myofibrillar disarray and myocyte cell death. Titin normally spans the half sarcomere from M-line to Z-line and functions as a spring, storing the energy during systole and diastole that helps restore the cell to resting length (see inset). Hence, titin is essential to cardiac function, and its degradation alters myocyte contractile function in several ways that could contribute to the clinical syndrome of heart failure.
At present, there is no generally accepted method to provide selective protection of the heart from damage induced by anthracyclines. Modifications in dosing schedule and administration of anthracyclines can make a significant impact on the incidence of heart failure, whereas modification of anthracycline structure can result in compounds with reduced cardiotoxicity and allow for higher dose administration (17). Co-administration of anthracyclines with antioxidants has been tried with limited success; the cardioprotective efficacy of antioxidants like vitamin E or N-acetylcysteine is only limited in large-sized animals such as dogs or pigs. Similarly, clinical studies fail to document mitigation of cardiomyopathy in patients given robust doses of vitamin E or N-acetylcysteine (18, 19). The iron chelator dexrazoxane reduces the incidence of contractile dysfunction in cancer victims treated with anthracyclines (20); however, the possibility that dexrazoxane reduces tumor response rates has prevented its universal employment (21). Current guidelines from the American Society of Clinical Oncology support the use of dexrazoxane in the patients who have received high doses of doxorubicin (over 300 mg/m2) during certain treatment regimes (22). Hence, in spite of the significant efforts to understand and prevent anthracycline-induced cardiotoxicity, there is no universally accepted strategy.
Mechanisms of Anthracycline Toxicity: Insights from Herceptin Therapy
Newer cancer therapeutics have been developed that target specific oncogene products involved in regulation of cancer growth. One of these therapeutics, trastuzumab (Herceptin), appears to increase the susceptibility of the heart to anthracyclines, and this experience provides some new insight into the mechanisms of anthracycline cardiotoxicity. Trastuzumab targets the erbB2 (HER2/neu) oncogene product, a receptor tyrosine kinase in the four-member erbB family that comprises erbB1 (i.e., EGFR), erbB2, erbB3, and erbB4. erbB2 is a non–ligand-binding 185-kD transmembrane protein with a cytoplasmic tyrosine kinase domain that is normally activated by heterodimerization with erbB1, erbB3, or erbB4 (23). ErbB2 is overexpressed in 25–30% of human breast cancers, and this overexpression is associated with enhanced tumor aggressiveness and a poor prognosis (24). Several monoclonal antibodies have been developed against the HER2 ectodomain to specifically inhibit the growth of tumor cell lines overexpressing HER2. One monoclonal antibody, 4D5 (25), was fully cloned, and the Fc portion was “humanized” to create trastuzumab (Herceptin) (26). Trastuzumab antitumor activity against HER2-positive human breast tumor cells in laboratory models led to clinical trials that ultimately demonstrated a clinical benefit in women with HER2-overexpressing breast cancers (27). On the basis of trastuzumab clinical efficacy, this antibody was approved in 1998 for treatment of patients with HER2-overexpressing metastatic breast cancer.
Significantly, a fraction of patients treated with trastuzumab develop clinical heart failure with a decrease in cardiac contractile function (28). This development was most notable in patients who concurrently received anthracyclines (Figure 3⇓); the incidence of cardiac dysfunction or symptomatic heart failure was about four times more likely in patients who received both trastuzumab and chemotherapeutic than in those who received anthracycline and cyclophosphamide alone. Although there was initial concern that the effect of trastuzumab on the heart would limit its use (29), more recent clinical trials have demonstrated lower rates of treatment-associated heart failure. Moreover, the treatment of those patients who develop heart failure with standard medical therapy suggests that the contractile dysfunction is often reversible (T. Suter, unpublished results). Nevertheless, this experience has triggered further research into the molecular mechanisms of anthracycline-induced cardiotoxicity that may lead to strategies for cardioprotection.
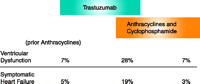
Trastuzumab (Herceptin) exacerbates chemotherapy-associated cardiotoxicity. Trastuzumab has antitumor activity against HER2-positive human breast tumor cells in laboratory models and is effective against human HER2-overexpressing breast cancers. In the clinical trials demonstrating the clinical benefit of trastuzumab in patients with metastatic breast cancer, there were an unexpectedly high number of patients who developed cardiac contractile dysfunction and clinically apparent heart failure. [From (28).]
Studies in animals and cell culture have provided some insight into the mechanisms for trastuzumab effects on the heart. Gene targeting studies in mice have demonstrated that erbB2, the target of trastuzumab, is essential for cardiac development (30), and conditional deletion of erbB2 in mice leads to the development of a dilated cardiomyopathy (31, 32). Thus, erbB2 is essential for maintenance of normal cardiac structure and function. In vitro studies with cardiac myocytes provide further insights regarding the cellular and molecular mechanisms for trastuzumab effects. erbB2 is expressed in adult cardiac myocytes, along with erbB4, and transmits growth and survival signals in response to the ligand neuregulin-1 (Figure 4⇓)(33). In response to anthracyclines, cardiac myocytes show evidence of myofilament degradation (12), as well as cell death by both apoptosis and necrosis (34), and both of these effects are suppressed by neuregulin treatment (35, 36). erbB2 and erbB4 localize in the transverse tubules of ventricular myocytes. Collectively, these data suggest that one role of erbB2/4 signaling is to dynamically regulate sarcomere structure, perhaps in response to stress. In this paradigm, suppression of erbB2 signaling by trastuzumab may accelerate the net breakdown of sarcomeric proteins induced by anthracyclines, increasing the likelihood of cardiac dysfunction and the clinical syndrome of heart failure.
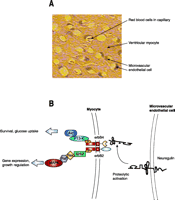
Paracrine signaling between cardiac endothelial cells and ventricular myocytes. A. As illustrated in this section of rat myocardium, ventricular myocytes are surrounded by microvascular endothelial capillaries. Besides supplying blood flow per se, the microvascular endothelial cells provide trophic support to the myocytes, which modulates cardiac structure and function. B. Beyond its role as a “glial growth factor,” neuregulin acts also as a paracrine signal between cardiac microvascular endothelial cells and ventricular myocytes. Neuregulin is expressed in the microvascular endothelial cell and protects myocytes from injury in the presence of anthracyclines and other cell stresses. Neuregulin activates erbB2 and erbB4 receptor tyrosine kinases in myocytes, thus regulating several downstream signaling cascades.
It is interesting that one of the most sensitive sarcomeric proteins is the giant myofilament protein titin (12). Titin plays a role in sarcomerogenesis, and its breakdown may impair sarcomere turnover (37). Moreover, titin plays a critical role in the regulation of contractile function, serving as an entropic spring imparting both a restoring force to assist in the return of the sarcomere to resting length during diastole, as well as a passive force to assist in the initiation of systole [for review see (38)]. Arguably, any cardiotoxic stress that accelerates the breakdown of titin will lead to a reduction in diastolic function, perhaps before any evidence of cardiac systolic (contractile) dysfunction.
Designing Cardioprotective Strategies
The observation that “turning down the volume” on erbB2 signaling with trastuzumab increases the likelihood of anthracycline cardiotoxicity leads to the hypothesis that augmentation of erbB2 signaling prior to or at the time of anthracycline exposure might be cardioprotective; certainly, in vitro studies with recombinant neuregulin support this hypothesis (35, 36). Recent work with the hemizygous neuregulin-1 (knockdown) mouse also supports this hypothesis (39). So, how can we “crank up the volume”?
Pharmacological strategies such as systemic delivery of neuregulin are one possibility, although the pleiotropic growth effects of this ligand, including in some cancers, makes this strategy seem less than optimal. Other pharmacological agents to consider along these lines include growth factors such as insulin-like growth factor-1 (IGF-1), which protect cells against anthracyclines (40). As is the case for neuregulin, however, there are concerns that any such factor may promote cancer growth. Obviously, the ideal strategy would be to locally deploy neuregulin and possibly other cardioprotective ligands in the heart or recruit downstream effector mechanisms. Intriguingly, overexpression of downstream effector kinases like Akt and transcription factors such as GATA4 protect mice from anthracycline cardiotoxicity (41, 42); however, these tools are still far away from clinical utility.
An attractive strategy yet to be explored is to use endogenous ligands and kinases to create a window of cardioprotection. Focusing on neuregulin, for example, we know that there is robust expression of this ligand in the heart, although it appears to have minimal activity at baseline. These levels of expression of neuregulin may well indicate that it serves some physiological function beyond cardiac development. If we knew the physiological mechanisms underlying neuregulin expression and activation in the heart, one could envision a scenario where the induction of neuregulin activity before exposing a patient to anthracycline would create a window of cardioprotection.
Along these lines, we have sought to characterize more fully the endogenous neuregulin expressed in the heart, and understand its mechanisms for activation. Neuregulins come from four known genes: NRG1, NRG2, NRG3, and NRG4. Cardiac neuregulin appears to be primarily a product of NRG1, the best characterized of these genes. The entire human NRG1 gene is 1.4 megabases long (1/2000th of the genome) (43). As a consequence of rich alternative splicing and multiple promoters, at least fifteen different NRG1 isoforms are produced (44). The three structural characteristics we know to differentiate isoforms with respect to in vivo functions and cell biological properties are the type of EGF-like domain (α or β), the N-terminal sequence (type I, II, or III), and whether the isoform is initially synthesized as a transmembrane or secretable protein. Cardiac microvascular endothelial cells transcribe at least eleven different NRG1 isoforms, including both α and β subtypes (G. Cote and D. Sawyer, unpublished data). All but one of these are predicted to be transmembrane proteins that require proteolytic cleavage for activation.
Gene targeting studies in mice demonstrate that both the EGF and the cytoplasmic domains are required for proper neuregulin function in cardiac development (45). In isolated cells, we have demonstrated that transmembrane neuregulin protein is cleaved in response to oxidative stress and is able to act on cardiac myocytes in a paracrine manner to induce cytoprotection (46). The cleavage of neuregulin under these conditions appears to require the action of a metalloproteinase, and work with recombinant neuregulin constructs has demonstrated a susceptibility of neuregulin to cleavage by ADAM17 and ADAM19 (a disintegrase and metalloproteinase) (47, 48) (Figure 4⇑).
Based upon experimental data in skeletal muscle, we hypothesize that physiologic cardiac stress in the form of exercise will increase neuregulin/erbB signaling. In skeletal muscle, neuregulin-1 and the erbB receptors serve a function at the neuromuscular junction to regulate myoblast differentiation as well as formation and maintenance of the post-synaptic endplate (48, 49). We have demonstrated that these proteins are localized in the adult skeletal muscle at the neuromuscular junction in rats (49,50). Moreover, we found that exercise was a potent activator of neuregulin release and subsequent activation of erbB phosphorylation. The proximal mechanisms for neuregulin release during exercise remain incompletely characterized. Interestingly, an exercise-training regimen in people increases the expression of erbB3 receptors in skeletal muscle (51). Increased erbB3 expression may thus increase sensitivity of muscle to neuregulin action. The demonstration that neuregulin increases skeletal muscle glucose uptake (52, 53) suggests the interesting hypothesis that exercise-regulated glucose uptake in skeletal muscle is mediated in part by neuregulin. (Regardless of the mechanisms for neuregulin release and the implications of these findings for glucose homeostasis, these studies have provided intellectual motivation for young neuregulin investigators to find the time to maintain their physical fitness.)
Coming back to the heart and anthracyclines, an attractive hypothesis that arises is that a period of exercise sufficient to activate neuregulin and/or other cardioprotective signaling may protect a cancer patient’s heart during anthracycline exposure. This makes intuitive sense, because periodic exercise is generally associated with cardioprotection and improvements in cardiovascular function (54– 55). This idea is not new, as twenty years ago a group demonstrated that swim training in rats decreased the histopathologic damage induced by anthracyclines (56). As far as we can see from the published literature, however, this concept was never pursued further, and cancer patients undergoing chemotherapy today remain without any clear recommendations with regards to physical activity around the time of treatment.
While we have arrived at this cardioprotective hypothesis via our studies of neuregulin and the insights from trastuzumab, there are many potential mechanisms through which physical activity may ameliorate anthracycline-induced cardiac toxicity. Kanter and colleagues have suggested that exercise training in rats acts to protect via changes in antioxidant activity. Specifically, they reported increased levels of catalase, superoxide dismutase, and glutathione peroxidase in blood, liver, and heart in swim-trained rats, and that this training prevented in part the cardiotoxicity of doxorubicin (56). Other potential mechanisms may involve the beneficial effects of exercise on expression of heat shock proteins (57–59), which are cardioprotective against anthracycline cardiotoxicity (60). Exercise effects on IGF-1 are well-characterized (61, 62), and this growth factor also induces protection from anthracycline exposure (63). Finally, the effects of exercise on adrenergic tone might also promote cardioprotection, based upon the finding that pharmacological administration of the alpha-adrenergic agonist phenylephrine is able to prevent anthracycline cardiotoxicity in mice (43). Two recent studies have confirmed the cardioprotective effect of exercise against doxorubicin toxicity (64, 65).
While the level of evidence for a protective effect of exercise on the heart during anthracycline exposure seems to be growing, concerns about the possible effects of exercise on tumor biology, response to chemotherapy, and patient tolerability remain. Extrapolating from epidemiologic data associating periodic exercise with a lower risk of the development of some forms of cancer, particularly breast and colon (66–69), one can argue that exercise does not promote tumor growth, although animal studies in breast cancer models have given conflicting data. Chemically induced breast cancers are suppressed by voluntary exercise in some settings (70, 71), but show the opposite results in others (72, 73). These conflicting data may be due to the tumor model as well as the form of exercise used in these studies. Regardless, further experimental data in oncogene-driven tumor models more applicable to human cancers should be helpful, as well as studies examining how exercise affects the tumor response to chemotherapy.
Cancer victims appear, in fact, to tolerate a regular physical activity program and report higher quality-of-life scores, suggesting the feasibility of an exercise intervention (74, 75). Unlike recombinant proteins and small-molecule activators of cardioprotective signaling, an exercise intervention comes without intellectual property rights, which of course is a disadvantage, as well as an advantage. Exercise therapy will not make any individual or institution wealthy, and carrying out this work will only get done with public and philanthropic funding to academic investigators. An advantage over patentable drugs, however, is that a proven exercise regimen that improves patient outcome is immediately exportable to all motivated cancer victims throughout the world, at no cost. This combination puts the investigator in the rather unique position of working on a therapy where the risk/benefit ratio is entirely driven by patient outcome, without financial conflicts of interest.
Acknowledgments
DBS is supported by the National Institutes of Health and acknowledges previous support from the American Heart Association, Juvenile Diabetes Research Foundation, Genentech, and Roche. CCL acknowledges funding from the American Heart Association.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Douglas Sawyer, MD, PhD, completed his medical and graduate education at Cornell University and his clinical training in Medicine and Cardiology at Brigham and Women’s Hospital. Doug is currently an Associate Professor of Medicine at Boston University Medical Center and is interested in the mechanisms by which paracrine signaling in the heart regulates cardiac structure and function. Address correspondence to DBS. E-mail Douglas. Sawyer{at}bmc.org; fax 617-414-1719.

Chee Chew Lim, PhD, has a background in bioengineering and completed his graduate work in Physiology at Boston University. Chee is an Assistant Professor of Medicine and studies the turnover of sarcomeric proteins in cardiomyocytes.

Billy Chen is a graduate student in the MD/PhD combined degree program at Boston Uniiversity School of Medicine. Billy’s thesis focuses on furthering our understanding of the mechanisms for anthracycline cardiotoxicity.

Xuyang Peng, MD, PhD, received her medical training at Hunan Medical School and clinical training in hematology and oncology at the Hunan Tumor Hospital in Hunan, China. Xuyang is currently completing a postdoctoral fellowship at Boston University Medical Center.

