CYP3A Probes Can Quantitatively Predict the In Vivo Kinetics of Other CYP3A Substrates and Can Accurately Assess CYP3A Induction and Inhibition
- 1Assistant Dean for Clinical Research, Professor and Research Director, Department of Anesthesiology; Adjunct Professor of Medicinal Chemistry, University of Washington School of Medicine, Seattle, WA 98195;
- 2Associate Dean for Research, Professor of Pharmaceutics, University of Washington School of Pharmacy, Seattle, WA 98195;
- 3Director, General Clinical Research Center, Verne S. Caviness Distinguished Professor of Medicine, University of North Carolina, Chapel Hill, NC 27599
The activity of cytochromes P450 (CYPs) can be a critical determinant of drug clearance, and interindividual variability in drug disposition and clinical efficacy. Changes in CYP activity underlie many drug interactions, often undesired. The use of select drugs as “probes” to assess in vivo CYP activity has been the subject of intense interest for over a decade (1), is advocated by numerous organizations, including the US Food and Drug Administration, European Federation of Pharmaceutical Sciences, the American Association of Pharmaceutical Sciences (2–4), and the pharmaceutical industry (5), and remains the subject of numerous ongoing investigations, papers, and editorials (6).
In a recent Viewpoint (7), Leslie Benet characterizes as failed all prior attempts to develop probes for the most clinically important P450, CYP3A. Moreover, he believes that useful CYP3A probes will never be developed and implies that further research in this area is futile. We disagree with this conclusion and challenge some of Dr. Benet’s interpretations of the data presented.
Benet correctly points out that CYP3A substrates can differ greatly in physical properties, which, in turn, result in differences in non-metabolic determinants of clearance including varying reliance on cellular uptake and efflux transporters. It logically follows that once all factors contributing to the clearance of a particular CYP3A probe are known, the “usefulness” of the probe is defined. However, Benet appears to argue that even CYP3A probes with defined metabolic and non-metabolic determinants of clearance do not provide useful information. To support this position, he reviews (in his Table 2) the data of Masica et al. (8) and Kharasch et al. (9, 10), where purportedly “poor correlations” were observed between structurally similar or dissimilar CYP3A probes in healthy adults with constitutive CYP3A activity (not receiving CYP3A inducers or inhibitors). The poorest correlations, however, were observed when one probe was administered orally and the other probe was administered intravenously. Because the activity of liver and intestinal CYP3A do not correlate (11–13), such poor correlations are expected.
The clinical usefulness of a relationship between the clearances of two drugs will depend on the coefficient of determination (r2), or other measure of association, as pointed out by Masica et al. (8) and Benet (7). The value of r2 is the product of biology (CYP3A activity and the true relationship between the clearances of two probes), but also of chemistry, intra-subject variability (if probes are administered on different days), and statistical considerations. These are unavoidable, and hence the expectation of r2 values near 1.0 is not tenable. Chemical (assay) variability is inherent in all drug concentration measurements, and as analytical methods improve, such variability will likely diminish. More important, however, is that sample size is a determinant of the precision of r2, and its statistical significance. Even with an excellent underlying correlation between two sets of biological data (assuming a normal distribution), a small sample size (< 20) may yield an imprecise estimate of the true r2, or fail to detect a significant correlation at all. Hence, it is not surprising that studies with small sample sizes have r2 values that do not achieve statistical significance. For example, in an investigation involving a sufficiently large sample size (n=95), the correlation between oral alfentanil and midazolam clearances in subjects with constitutive CYP3A activity was highly significant (r2 = 0.60, p< 0.001), indicating >50% of the variability in clearance of one probe was predicted by the clearance of the other (14). When, however, small sample sizes were evaluated (although such analyses were not reported in the original publications because the studies were not powered for such), the r2 values were smaller and more variable (n=9, r2= 0.52, n=18, r2= 0.17) (9, 15), as correctly pointed out by Masica et al. (8) and Benet (7). Therefore, a robustly powered study was needed to reveal the true magnitude of a biological association, presumably reflecting the negative impact of sample size on the precision of the measured r2 value.
One issue regarding CYP3A probe utility is its ability to detect induction and inhibition. Even more stringent than simply detecting a qualitative change, is whether the magnitude of change in the clearance of one probe predicts that of another. Whereas Benet suggests that evaluating the predictive accuracy of a CYP3A probe under conditions of induction and inhibition is somehow inappropriate, we disagree with this opinion. Any test must be validated over the intended range of use. This applies to laboratory assays (i.e., standard curves) as well as clinical tests. If alfentanil (or any other CYP3A probe) is to be used to assess CYP3A induction or inhibition, it must be, and was, validated over the range of anticipated clinical CYP3A activities (9, 10). Moreover, such validation strengthens its use in assessing subjects with CYP3A at the extremes of activity, independent of a drug interaction. Indeed, in the investigation cited by Benet, the ratios (treated/control) of area under the concentration vs time curves (a quantitative measure of CYP3A induction or inhibition) for alfentanil and midazolam were highly correlated (Figure 1⇓) (10). Thus, the available data suggest that estimates of the magnitude of induction or inhibition of CYP3A activity are essentially identical with either probe, a CYP3A probe can quantitatively predict the in vivo kinetics of other CYP3A substrates, and can accurately assess CYP3A induction and inhibition. More research is needed to assess whether the quantitative predictive accuracy of alfentanil or midazolam can be widely generalized. Nonetheless, it is not reasonable to assert that every combination of clinically used inducers and inhibitors must actually be tested.
A second issue regarding CYP3A probe utility is its ability to quantitatively assess CYP3A activity in subjects with constitutive CYP3A activity, perhaps with the goal of guiding the dosing of another CYP3A substrate. We believe it is quite premature to declare that no CYP3A probe will ever be able to predict enzyme activity in an individual subject (8), or that further investigations in this arena will not be productive (7). Indeed, as highlighted by Masica et al. (8), “in contrast to a widely held perception, the head-to-head comparison of one CYP3A in vivo probe to predict that of another under constitutive conditions has not, in fact, been extensively investigated”. Nevertheless, as illustrated above, if sample sizes are considered appropriately, one CYP3A probe (alfentanil) can quantitatively predict one-half of the variability in the in vivo kinetics of another CYP3A substrate (midazolam), in subjects with constitutive CYP3A activity.
Identification of an in vivo probe for P-glycoprotein (P-gp), or other transporters, is in earlier stages of development, in large part because our knowledge of basic transporter biology is less complete than that of CYP. The primary purpose of the paper by Kharasch et al. (16) was to demonstrate that a probe for CYP (alfentanil, which is not known to be a transporter substrate) and a probe for P-gp (fexofenadine, which is not metabolized by CYP3A) could be administered together without significant pharmacokinetic interaction. Use of fexofenadine as a P-gp probe had been proposed previously by others (17–19), and Kharasch et al. (16) did clearly specify that it may be a substrate for organic anion-transporting polypeptides as well. Recently, it has been proposed that the erythromycin breath test can be modified to separately measure CYP3A4 and P-gp activities in liver and intestine, capitalizing on the roles of metabolism and transport of this substrate (20). As transporters and their substrates are still being continuously identified, abandoning the assessment of their activity in vivo certainly seems premature.
As identified above, the use of probes to assess in vivo CYP activity has applicability towards drug development, and has regulatory endorsement, if not requirement. Although in vitro systems exist for detecting CYP inhibition, and, more recently, CYP induction, in vivo verification is necessary. And certainly the absence of an interaction with an in vivo probe is very predictive.
A final issue is the utility of CYP or transporter probes in the clinic. It is accepted that interpatient variation in drug pharmacokinetics is a reason for therapeutic failure or toxicity from many drugs. Patients are not healthy volunteers and variation in CYP or transporter activity may be much greater in patient populations owing to the effects of medications and concomitant disease. Therefore, data obtained in healthy volunteers may not be applicable to all clinical situations. Moreover, probes do not have to give precise quantitative information to be clinically useful given the complete knowledge vacuum often confronting the prescribing physician. A probe that predicts 50% of interpatient variation in elimination kinetics may be useful, compared to current largely empirical dosing paradigms, for dosing a drug with a narrow therapeutic index (6, 21).
In summary, the idea that the clearance of any CYP3A probe will accurately predict the clearance of all other CYP3A substrates was as naive as the contention that antipyrine would predict clearance of all drugs metabolized by “cytochrome P450.” More research is needed to define further the nonmetabolic factors underlying clearance of CYP3A substrates. In the meantime, CYP3A probes continue to provide useful information in research and drug development, and will likely find clinical utility.
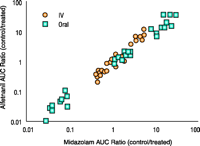
One CYP3A probe (alfentanil) can predict the disposition of another CYP3A substrate (midazolam). Data are the ratios (treated/control) of area under plasma concentration–time curve extrapolated to infinity for oral and IV alfentanil and midazolam, in subjects treated with rifampin (hepatic and intestinal CYP3A induction), troleandomycin (hepatic and intestinal CYP3A inhibition), or grapefruit juice (intestinal CYP3A inhibition). Data are replotted from Kharasch et al. (10).
Footnotes
-
Note: Paul B. Watkins is equity owner in Metabolic Solutions, Inc, Nashua New Hampshire that manufactures the erythromycin breath test.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Paul B. Watkins, MD, is the Verne S. Caviness Distinguished Professor of Medicine and Director of the General Clinical Research Center at the University of North Carolina, Chapel Hill. His research focuses on the molecular determinants of interpatient variation in drug pharmacokinetics and susceptibility to hepatotoxicity. Please address Email correspondence to PBW at pbwatkins{at}med.unc.edu

Ken Thummel, PhD, is Professor of Pharmaceutics and Associate Dean for Research and New Initiatives in the School of Pharmacy at the University of Washington, Seattle, WA. His research focuses on mechanisms of inter-individual variability in drug elimination processes and CYP3A-dependent intestinal first-pass metabolism. Please address E-mail correspondence to KT at thummel{at}u.washington.edu

Evan D. Kharasch, MD, PhD, is Professor of Anesthesiology and Director of Anesthesiology Research, and Assistant Dean for Clinical Research in the School of Medicine, at the University of Washington, Seattle, WA. His research interests include pharmacokinetic and pharmacokinetic drug interactions and interindividual variability, methods of assessing CYP3A activity, and mechanisms of anesthetic toxicity. Please address E-mail correspondence to EDK at kharasch{at}u.washington.edu; fax 206-685-3079.

