ROLE of MITOCHONDRIA in TOXIC OXIDATIVE STRESS
- 1Department of Pharmaceutical Sciences, 2University of Colorado Cancer Center, University of Colorado Health Sciences Center, Denver, CO 80262, USA;
- 3Department of Biomedical Sciences, University of Prince Edward Island, Charlottetown, PE Canada C1A 4P3;
- 4Program in Neuroscience, University of Colorado Health Sciences Center, Denver, CO 80262, USA;
- 5Laboratory of Molecular Genetics, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA;
- 6Institute of Environmental Medicine, Division of Toxicology, Karolinska Institutet, SE-171 77 Stockholm, Sweden
Abstract
Oxidative stress and mitochondrial oxidative damage have been implicated in the etiology of numerous common diseases. The critical mitochondrial events responsible for oxidative stress–mediated cell death (toxic oxidative stress), however, have yet to be defined. Several oxidative events implicated in toxic oxidative stress include alterations in mitochondrial lipids (e.g., cardiolipin), mitochondrial DNA, and mitochondrial proteins (eg. aconitase and uncoupling protein 2). Furthermore, recent findings indicate the enrichment of mitochondrial membranes with vitamin E protects cells against the toxic effects of oxidative stress. This review briefly summarizes the role of these mitochondrial events in toxic oxidative stress, including: 1) the protective role of mitochondrial vitamin E in toxic oxidative stress, 2) the role of mitochondrial DNA in toxic oxidative stress, 3) the interaction between cardiolipin and cytochrome c in mitochondrial regulation of apoptosis, 4) the role of mitochondrial aconitase in oxidative neurodegeneration, and 5) the role of mitochondrial uncoupling protein 2 in the pathogenesis of type 2 diabetes.
Introduction
The primary role of mitochondria is the generation of ATP through oxidative phosphorylation (OXPHOS) and oxygen consumption. Mitochondria possess both an outer and inner membrane, the latter of which has a larger surface area, is impermeable to all molecules, and contains the enzymes responsible for oxidative phosphorylation. Mitochondrial ATP production occurs through the flow of electrons derived predominately from intermediates of the tricarboxylic acid cycle such as nicotinamide adenine dinucleotide (NAD) or flavin adenine dinucleotide (FAD) reducing equivalents (Figure 1⇓). These electrons are passed along a series of molecular complexes in the inner mitochondrial membrane known as the electron transport system (ETC). As a result of this electron transfer, protons are transferred across the inner mitochondrial membrane producing a large mitochondrial membrane potential (Figure 1⇓). Energy lost by protons re-entering the mitochondrial matrix through F0F1 ATP synthase is used to produce ATP. The final electron acceptor is molecular oxygen, which is reduced to water.
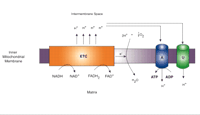
Schematic representation of mitochondrial electron transport chain (ETC) and coupling and uncoupling of oxidative phosphorylation. Reduced substrates NADH and FADH2 are oxidized, with electrons passing to the enzyme complexes of the ETC and the protons (H+) pumped into the intermembrane space of the mitochondrion, forming a large mitochondrial membrane potential known as the proton-motive force. Energy lost by protons re-entering the mitochondrial matrix through ATP synthase (A) is used to power generation of ATP from ADP. Protons may also re-enter the matrix via uncoupling proteins (U), such as UCP2, without production of ATP.
During the transfer of electrons along these electron transport complexes, single electrons sometimes escape and result in a single electron reduction of molecular oxygen to form a superoxide anion (O2·−). It is estimated that as much as 1% of all oxygen consumed may result in the formation of reactive oxygen species (ROS) such as superoxide anions, with the vast majority generated in mitochondria (1). Furthermore, this mitochondrial superoxide production can be significantly enhanced if the rate of electron transport is limited by the buildup of a large proton gradient in the inner mitochondrial membrane (IMM). Such a proton buildup can occur with abundant fuel supply (NADH production) or with the functional impairment of one or more of the electron transport complexes (especially complexes I and III) (2). Mitochondrial O2·− generation can then lead to the production of ROS such as hydrogen peroxide (H2O2), which can, in the presence of ferrous iron via the Fenton reaction, result in highly reactive hydroxyl radicals. Once mitochondrial enzymatic and nonenzymatic antioxidant systems are overwhelmed by these ROS, oxidative damage and cell death can occur.
Although energy production is an important function of mitochondria, these organelles also participate in initiating and executing both apoptotic and necrotic cell death as well as in maintaining calcium and iron homeostasis (3). Thus, it appears that mitochondria serve as a central barometer for assessing changes in cellular viability. Mitochondrial-related events such as a decline in OXPHOS (ATP production), the onset of mitochondrial permeability transition, increased concentrations of free Ca2+ and bioavailable ferrous iron, and oxidative damage to key mitochondrial constituents appear to be important contributors to the demise of the cell, either collectively or individually (4–7). Based on our present knowledge of mitochondrial function, it is not surprising that mitochondrial oxidative stress and mitochondrial oxidative damage have been implicated in the etiology of numerous common diseases. These prominent clinical conditions include liver disease [e.g., alcoholic, iron overload, or nonalcoholic fatty liver], ischemia–reperfusion injury, diabetic complications, neuro-degenerative diseases, cancer, aging, and many others (8–12). Though the critical mitochondrial event(s) responsible for oxidative stress-mediated diseases have yet to be defined, experimental and clinical evidence support oxidative damage to mitochondrial lipids, nucleic acids, and proteins as important events in these toxic processes. Furthermore, the enrichment of mitochondria with antioxidants has been shown to protect cells against toxic oxidative stress. Clearly, protective strategies applicable to these deadly clinical conditions could be successfully developed with a better understanding of the critical molecular events in mitochondria that are responsible for toxic oxidative stress.
The present review summarizes evidence for the critical role of specific mitochondrial events such as damage to mitochondrial DNA; to the mitochondria-specific lipid, cardiolipin; to mitochondrial aconitase; and the induction of mitochondrial uncoupling protein 2 in toxic oxidative stress. In addition, the role of mitochondrial vitamin E in preventing mitochondrial oxidative damage and toxic injury is also discussed.
Protective Role of Mitochondrial Vitamin E in Toxic Oxidative Stress
Because mitochondrial oxidative damage plays an important role in the etiology of numerous oxidative stress–mediated clinical conditions, one possible protective strategy would be to enrich tissue mitochondria with antioxidants thereby limiting mitochondrial oxidative damage, cellular injury and the initiation and progression of disease. This strategy has been employed successfully in recent in vitro and in vivo experiments using the administration or induction of enzymatic and nonenzymatic antioxidants that are targeted to mitochondria. For example, the two enzymatic antioxidants mitochondrial phospholipid hydroperoxide glutathione peroxidase (PHGPx) and Mn-superoxide dismutase (MnSod, or Sod2) dramatically reduce, respectively, lipid hydroperoxide and superoxide anion levels in mitochondria (13, 14). Both enzymes, upon induction or overexpression in cells, effectively limit toxic oxidative stress in vitro and in vivo (15, 16). The overexpression of nonmitochondrial PHGPx has often failed, however, to prevent cell death induced by these same oxidative insults (13). Data from these previous studies support the concept that mitochondrial oxidative damage is a critical event responsible for toxic oxidative stress and that the enrichment of mitochondria with antioxidants can protect this organelle and the cell from toxic injury. The early reports on the cytoprotective ability of mitochondrial enzymatic antioxidants have led to the recent synthesis of nonenzymatic antioxidants that are targeted to mitochondria. To deliver these synthetic antioxidants selectively to mitochondria, investigators (17) have attached a lipophilic triphenylphosphonium cation to alpha tocopherol (MitoVit E) or to ubiquinone (MitoQ). The positive charge of these antioxidant derivatives enables them to accumulate several hundred-fold within mitochondria because of the negative membrane potential inside mitochondria. Cell culture studies using these mitochondria-targeted antioxidants have demonstrated the protective abilities of these molecules against toxic oxidative stress (17, 18). Other mitochondria-targeted antioxidants include synthetic peptides (SS) that selectively concentrate in the inner mitochondrial membrane (19). These peptides scavenge ROS at the site of production thereby limiting mitochondrial oxidative damage and cell death. Using these cytoprotective peptides, investigators have shown that mitochondrial oxidative stress and damage are responsible for hydrogen peroxide– and 3-nitropropionic acid–induced cell death as well as myocardial ischemia–reperfusion injury (19). Though mitochondria-directed antioxidants are in the early stage of development, they hold great promise as a tool to define the critical mitochondrial events responsible for toxic oxidative stress as well as a potential therapy for the treatment of diseases associated with oxidative stress.
Previous studies have also demonstrated that the enrichment of hepatic mitochondria with endogenous d-alpha tocopherol (T) is critical for cytoprotection against toxic oxidative stress (11, 20–22). Thus, the delivery of endogenous T to mitochondria also holds promise as a cytoprotective strategy because T is the predominate membrane-bound antioxidant in mammalian cells that scavenges oxygen radicals and protects lipids, nucleic acids, and proteins against oxidative damage (23, 24). This chain-breaking antioxidant is not synthesized by mammalian cells, and once membrane T is consumed (oxidized to inactive form) during periods of oxidative stress, cellular macromolecules are subject to peroxidation that can result in toxic injury (25, 26). Surprisingly, the T content in cellular membranes and especially mitochondrial membranes, is maintained at low levels and close to the threshold concentration required to halt the propagation of lipid peroxidation and the oxidation of cellular proteins and nucleic acids (11, 27–29). Mitochondrial membranes possess a limited amount of T that is not easily replenished, thus, a viable protective strategy against toxic oxidative stress appears to be the enrichment of mitochondrial membranes with endogenous T.
The concept that enrichment of mitochondria with protective levels of T is critical for preventing toxic oxidative stress originated from previous studies demonstrating that the treatment of hepatocytes with the succinate ester of T (TS) treatment protected these cells from a wide variety of toxic oxidative challenges (30) (Table 1⇓). Interestingly, numerous investigators have reported that TS is far more effective than T or d-alpha tocopheryl acetate (TA) in protecting normal cells against many different acute toxic oxidative challenges including mitochondrial toxicants (e.g., alkylating agents, iron, hyperoxia, doxorubicin and rotenone) (11, 21, 26, 31–33). In agreement with these studies, cellular toxicity observed following incubation with the toxic agents listed in Table 1⇓ were not prevented by pretreating hepatocytes with equimolar levels of T or TA, in contrast to TS treatment. These findings lead to the conclusion that TS has unique cytoprotective properties that may intervene in a common final pathway to cell death (26, 30, 33). Accordingly, defining the mechanism of TS-mediated cytoprotection may result in a better understanding of the critical molecular events that are responsible for toxic oxidative stress. Following considerable study, a mitochondria-related mechanism for the superior cytoprotective action of TS as compared with other vitamin E analogs has recently been demonstrated. Both in vitro and in vivo studies confirm that TS acts as a unique mitochondrial delivery system for T, rapidly accumulating in cellular and mitochondrial membranes and releasing active T to prevent mitochondrial oxidative damage and oxidative stress-induced cell death (11, 21, 22). By comparison, the acute administration of T or TA does not appear to enrich mitochondria with T nor protect hepatocytes from toxic oxidative stress (11, 21). For example, in hepatocytes incubated with 25 μM TS for sixty minutes, these cells and their mitochondria were completely protected from Fe2+- induced lipid peroxidation (as determined by an antioxidant capacity assay) and the hepatocytes were completely protected against oxidative stress-mediated cell death (21). In contrast, the protection of mitochondria and cells were not observed following three hours of T or TA treatments (21). From these previous studies, investigators have shown that TS cytoprotection is dependent on the uptake of the intact anionic-charged TS molecule, on the hydrolysis of TS to T, on the maintenance of mitochondria ultrastructure, and on the inhibition of mitochondrial lipid peroxidation [even when cellular and mitochondrial stores of glutathione (GSH) and ATP were depleted] (11, 21, 22). Importantly, similar findings have been observed following the acute in vivo administration of TS whereby rapid enrichment of hepatic mitochondria with TS and T protects against oxidant-induced mitochondrial permeability transition and oxidative stress-induced necrotic cell death (not observed with T or TA administration). For example, a single intraperitoneal (ip) injection of T or TS resulted in a significant increase (3–5 fold) in liver homogenate T levels (eighteen hours after administration), providing these membranes with complete protection against Fe2+- induced lipid peroxidation (11, 21). Only the administration of TS, however, resulted in a significant enrichment (3-fold) of hepatic mitochondrial membranes with T, which again completely prevented Fe2+-induced lipid peroxidation and cell death (11, 21). Isolated hepatic mitochondria and hepatocytes from T- or TA-treated rats were not protected (11, 21). These results support a selective mitochondrial uptake process for this anionic T derivative. The TS molecule itself does not possess antioxidant properties (34), thus, the abundance of active unesterified T in liver homogenate and mitochondria isolated from TS-treated rats indicates the presence of esterase activity in both hepatocytes and their mitochondria to release sufficient T to protect these membranes against toxic oxidative damage. Unfortunately, we lack a basic understanding of the uptake and hydrolytic processes that are responsible for TS cytoprotection, and additional studies are needed. Both in vitro and in vivo results, however, confirm that TS acts as a unique delivery system for T, rapidly accumulating in cellular and mitochondrial membranes and gradually releasing active T to prevent mitochondrial membrane oxidative damage and cell death. These findings suggest that TS administration may prove useful for the prevention and treatment of oxidative stress-mediated diseases, especially those of mitochondrial origin. As TS is extensively hydrolyzed in the gut upon oral administration, parenteral administration of TS is required for the cytoprotective properties described above.
Protective Effect of Tocopheryl Succinate in Hepatocytes Exposed to a Variety of Toxic Oxidative Insults
As concluded from the TS protection studies detailed above, the enrichment of mitochondria with protective endogenous T appears to be an excellent therapeutic strategy for preventing toxic oxidative stress. Rather than using parenteral TS administration to rapidly supplement mitochondria with endogenous T, a more simplistic enrichment strategy might be dietary vitamin E supplementation, which is readily available both experimentally and clinically. Although considerable information is available concerning the tissue uptake and plasma transport of T following oral administration (35, 36), our knowledge of the regulation of subcellular T transport or, more specifically, the factors that promote the accumulation of T in mitochondria are limited. It is well know, however, that T rapidly accumulates in the liver following dietary supplementation, and thus initial studies were conducted to determine the effect of dietary vitamin E supplementation on enrichment of T in hepatic mitochondria and on cytoprotection against toxic oxidative stress. Male Sprague Dawley rats were fed a typical daily rodent diet that contained 50 (control), 250, or 1400 IU TA/kg (equivalent in humans to the daily consumption of approximately 40 mg, 200 mg, or 1000mg vitamin E). After seven days on these diets, animals were sacrificed and mitochondria isolated from 1400 IU TA diet–treated rats showed a dramatic (eight-fold) increase in T levels in both the liver homogenate and IMM (as compared to those in the 50 IU– or 250 IU–treated rats) as well as complete protection against Fe2+-induced lipid peroxidation (11) (Figure 2⇓). In contrast, liver homogenates and IMM isolated from 50 IU-treated rats were highly susceptible to lipid peroxidation (11). Interestingly, the 250 IU TA diet resulted in a two-fold T enrichment of liver homogenate that resulted in complete resistance to lipid peroxidation but only modest T enrichment in IMMs and modest cytoprotection (11) (Figure 2⇓). Thus, only IMMs and hepatocytes isolated from rats fed a diet containing 1400 IU TA were completely protected (identical to that observed for parenteral TS treatment with 125 mg/kg body weight, ip) against toxic oxidative insults such as iron overload (Figure 2⇓). These results demonstrate that dietary vitamin E supplementation can enrich hepatic mitochondria with protective levels of T in a dose-dependent manner and suggest that a greater than three-fold increase in hepatic T levels are required for the protective enrichment of mitochondria and IMMs. We conclude that dietary vitamin E supplementation may prove beneficial for the treatment of oxidative stress–mediated liver diseases, especially those of mitochondrial origin. Identifying whether other tissues, in addition to the liver, can rapidly enrich their mitochondria and IMM with protective amounts of T following dietary supplementation with vitamin E will require additional studies.
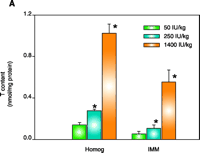
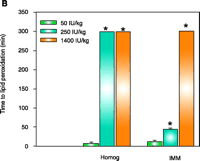
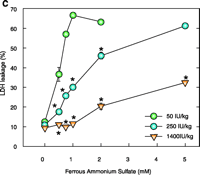
Effect of dietary TA supplementation on (A) T levels, on (B) susceptibility to lipid peroxidation in liver homogenates and inner mitochondrial membranes (IMM), or on (C) Fe2+-induced cell death. Male Sprague Dawley rats were given the diets that included TA supplements (50, 250, or 1400 IU/kg diet) for seven days. Liver homogenates (Homog) and IMMs or hepatocytes were isolated and examined by methods described elsewhere (21). Fe2+ -induced cell death was measured after two hours. Leakage of lactate dehydrogenase (LDH) from cells used as a measure of necrotic cell death. Data are the mean ± SE of three separate animals per treatments. *p<0.05, as compared with 50 IU/kg diet group. Figure adapted from (11).
In conclusion, experimental evidence clearly supports a protective role for mitochondrial vitamin E enrichment in toxic oxidative stress. The use of exogenously administered T to enrich mitochondria holds great promise in experimental systems to define the critical mitochondrial events responsible for toxic oxidative stress as well as in the treatment of diseases associated with oxidative stress.
Role of Mitochondrial DNA in Toxic Oxidative Stress
During the last three decades, numerous human diseases have been associated with the generation of ROS and the ensuing mitochondrial dysfunction that results in cell death [see (37) for review]. ROS can damage macromolecules within the mitochondria, including lipids, proteins, and nucleic acids. In human cells, each mitochondrion has approximately fifteen copies of a small genome consisting of 16,569 base pairs (Figure 3A⇓). This mitochondrial DNA (mtDNA) encodes thirteen polypeptides, twenty-two transfer RNAs (tRNAs), and two ribosomal RNAs (rRNAs), all of which are essential for electron transport and ATP generation, and consequently for normal cellular physiology. MtDNA, therefore, represents a critical cellular target for oxidative damage that could lead to lethal injury through the loss of electron transport, mitochondrial membrane potential, and ATP generation.
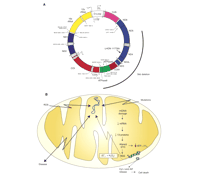
Mitochondrial DNA and oxidative stress. A. Mitochondrial genome from human cells indicating the genes, and some mitochondrial mutations associated with human disease. Mitochondrial DNA Map http://www.gen.emory.edu/mitomap.htmlB. Role of mtDNA in oxidative stress–induced injury. MtDNA damage can lead to loss of expression of mitochondrial polypeptides, subsequent decrease in electron transport and increased generation of reactive oxygen species (ROS), loss of mitochondrial membrane potential, and release of signals for cell death, such as cyt c and apoptosis-inducing factor (AIF).
In addition to mitochondria’s role in energy production and apoptosis, these organelles serve as sites of iron-sulfur cluster synthesis. Iron, like oxygen, is essential for life, but together they can form a witch’s brew of highly ROS. A yeast model exists of the human disease Friedreich’s ataxia (FA), which is characterized by decreased expression of the iron-binding protein frataxin (38–40). Yeast represents an important model system that can be easily challenged by genetic manipulation and offers a wide array of bio-informatics tools that are not available for the study of rodents or humans. Frataxin is located in the mitochondria, and its progressive loss in a yeast frataxin homolog 1 (yfh1) mutant leads to a rapid increase in mitochondrial iron, oxidation of proteins, and mitochondrial, as well as, nuclear DNA damage (41). The final outcome of the lack of frataxin in yeast is the generation of petite cells that lack mtDNA (rho0, respiration-deficient) and are, thus, defective for OXPHOS. The cellular impact of excessive mitochondrial iron in the yeast FA model was determined by global gene expression profiling of a strain with reduced levels of frataxin as compared to a rho0 strain. High concentrations of iron rapidly reduce the transcription of a large set of genes that encode mitochondrial ribosomal proteins, as well as, reduce OXPHOS and the total complement of electron transport proteins. This dysregulation of electron transport can lead to increased mitochondrial generation of H2O2. The combination of increased iron and physiological concentrations of H2O2 generated in the mitochondria during OXPHOS might result in the generation of DNA damage. Thus, accumulation of mtDNA damage and subsequent decline in mitochondria function could be a primary cause in the disease course of FA.
In yeast cells, as in mammalian cells, mtDNA is much more sensitive than nuclear DNA (nDNA) to H2O2-induced damage, possibly owing to iron availability in mitochondria. Also, human cells have a limited capacity for repairing H2O2- induced mtDNA damage (42). In order to understand the molecular events that follow oxidative stress and lead to persistent lesions in mtDNA, the formation and repair of H2O2-induced DNA lesions in telomerase-expressing normal human fibroblasts (NHF hTERT) were examined using quantitative PCR (QPCR) (43). The rationale of this assay is that any polymerase-blocking lesion leads to a decrease in DNA amplification (44), thus, DNA damage is inversely proportional to the amount of amplification. Because this assay is not dependent upon isolation of mitochondria and purification of mtDNA away from nuclear DNA, the assay is not prone to oxidation artifacts associated with HPLC electrochemical detection of 8-oxo-deoxyguaninosine and 8-oxo-guanine (45, 46). After exposure to 200 μM H2O2, NHF hTERT-expressing cells show extensive mtDNA damage (~4 lesions/10 kb), which is partially repaired during a recovery period of six hours post-exposure. At the same time, the nDNA seems to be completely resistant to damage. Cell-sorting experiments reveal persistent mtDNA damage twenty-four hours post-exposure, but only in the fraction of cells with low mitochondrial membrane potential. Further analysis also revealed increased production of H2O2 by these cells, which subsequently undergo apoptosis (43). This work supports a hypothesis for a feed-forward cascade of ROS generation and mtDNA damage and also suggest a possible mechanism for persistence of lesions in the mtDNA involving a drop in ΔΨm, compromised protein import, secondary ROS generation, and loss of repair capacity.
Seven differently derived sets of human fibroblasts or epithelial cells immortalized with hTERT suffered more H2O2-induced mtDNA damage than did normal diploid parental cells (47). hTERT expression neither altered the rate of H2O2 breakdown nor altered the endogenous amount of H2O2 in cells. Because the damaging effects of H2O2 are mediated by divalent metal ions (Fenton chemistry), the amounts of cellular, bioavailable metals were examined. In all cases, higher concentrations of chelatable metals were found in hTERTexpressing cells. In addition, the amount of hTERT expression was directly correlated with increased bioavailable iron, suggesting that the increased mtDNA damage likely arises from increased mitochondrial iron and associated Fenton chemistry.
Analysis of the hTERT amino-acid sequence revealed a putative mitochondrial targeting sequence. Confocal microscopy imaging of a hTERT–green fluorescent protein (GFP) fusion protein demonstrated mitochondrial localization of hTERT. Telomeric-repeat amplification protocol (TRAP) analysis of mitochondrial extracts indicated that mitochondria contain telomerase activity (47). Taken together, these two studies suggest that increases in mitochondrial iron, either through improper homeostasis by a decrease in frataxin or by ectopic hTERT expression, can lead to increased oxidation damage to mtDNA. In addition, these results suggest that mitochondrial telomerase sensitizes cells to oxidative stress, which can lead to apoptotic cell death, and imply a new function for telomerase in mitochondrial DNA. Finally, these results indicate that mtDNA damage following oxidative stress can lead to a vicious cycle of ROS propagation and mtDNA oxidation. The cascade initiated by oxidative mtDNA damage that leads to faulty gene expression, deficiency of key electron transport enzymes, subsequent ROS generation and ultimately, cell death, is known as the mitochondrial catastrophe hypothesis (Figure 3B⇑). MtDNA damage, therefore, represents an important target for intervention and as a biomarker in the course of many human diseases.
Cardiolipin-CytochromeC Interaction in Mitochondrial Regulation of Apoptosis
Apoptosis is a genetically controlled and evolutionarily conserved form of cell death essential for normal embryonic development and for the maintenance of tissue homeostasis in the adult organism. Apoptotic cell death is triggered by receptor-mediated (extrinsic) and mitochondrially-mediated (intrinsic) signaling pathways. In certain cell types, the receptor-mediated pathway of apoptosis recruits the mitochondrial death machinery to amplify the apoptotic response.
The release of different proteins from the intermembrane space of the mitochondria is a critical early event in mitochondrially mediated apoptotic cell death (3). Cytochrome c is one such protein which, upon extrusion into the cytosol, forms a complex with apoptotic protease-activating factor 1 (Apaf-1) and procaspase-9, resulting in the activation of the caspase cascade which brings about most of the biochemical and morphological alterations characteristic of apoptosis.
The detailed mechanisms of cytochrome c release from mitochondria remain elusive. Indeed, studies have focused more on the permeabilization of the outer mitochondrial membrane by either permeability transition pore (PTP) opening or the action of pro-apoptotic members of the Bcl-2 family of proteins, such as Bid, Bax, and Bak (48). Both processes result in cytochrome c release, but the underlying mechanism for cytochrome c mobilization from its binding to the inner mitochondrial membrane, which is a pre-requisite for its release, has been virtually ignored.
Cytochrome c is a water-soluble, basic, heme-containing protein that binds to the phospholipid cardiolipin (CL) located exclusively on the IMM of eukaryotic cells (49). Under normal physiological conditions, CL anchors cytochrome c protein to the mitochondrial membrane where it participates in electron transport between complexes III and IV of the respiratory chain; anchorage to the IMM ensures a limited soluble pool of cytochrome c.
CL is an anionic phospholipid whose unique structure among phospholipids confers fluidity and stability to the mitochondrial membrane [see (50) for review]. The molecular interaction between CL and cytochrome c involves electrostatic interactions at the A-site of cytochrome c, whereas hydrophobic interactions and hydrogen bonding take place at its C-site (49). In other words, one of the acyl chains of CL is inserted into a hydrophobic channel in cytochrome c while another acyl chain from CL extends into the phospholipid bilayer. This tight interaction is demonstrated spectroscopically by the heme-iron spin-state.
In accordance with these findings, Ott et al. (51) proposed that the electrostatic and hydrophobic interactions between CL and cytochrome c must be “breached” in order for cytochrome c to leave mitochondria. This hypothesis is also supported by previous studies demonstrating that the peroxidation of CL virtually abolishes cyto-chrome c binding (52). Furthermore, early studies utilizing bovine heart submitochondrial particles showed that mitochondrially generated ROS decreased the content of CL in IMM, which was concomitant with a decrease in the activity of cytochrome c oxidase (53). CL content was restored with exogenously added CL, but not with peroxidized CL, and the entire system was protected by the addition of the antioxidant enzymes, superoxide dismutase and catalase.
Unlike other phospholipids, the fatty acyl groups on CL are essentially restricted to C18 chains. The dominant C18 chain in mammals is linoleoyl (18:2), with oleoyl (18:1), and linolenoyl (18:3) also present. The unsaturated nature of the acyl chains of CL confer functional specialization and appear to be required for optimal function of many of the proteins and enzymes responsible for mitochondrial energy metabolism. The degree of unsaturation of the acyl chains is also important for the optimal synthesis of CL. Palmitate, a free, saturated (16:0) fatty acid found in plasma at high concentrations during ischemia and reperfusion, is known to induce apoptosis in many cell types. Importantly, palmitate-induced apoptosis in rat neonatal cardiomyocytes has been attributed to a decrease in CL synthesis, which correlates almost stoichiometrically with the extent of cytochrome c release into the cytosol (54). These findings are consistent with those of Hardy et al. (55), who found that palmitate (16:0) increased apoptosis (with concomitant cytochrome c release) whereas oleate (18:1) supported proliferation of breast cancer cells. Hence, it appears that palmitate induces CL turnover and decreases the concentration of fatty acid precursors needed for optimal CL synthesis, thereby reducing overall amounts of CL .
Pro-apoptotic Bcl-2 family proteins, which normally reside in the cytosol of mammalian cells, can induce the release of cytochrome c from isolated mitochondria (Figure 4⇓). This well-established finding has led to a search for membrane constituents that are responsible for attracting these proteins to the mitochondria. Model systems of synthetic liposomes, isolated mitochondria, and Chinese hamster ovary cells were used to demonstrate that CL is required for targeting tBid (truncated Bid, the active form of the protein) to mitochondria (56). Furthermore, the extent of tBid binding depends on the concentration of CL in both liposomes and mitochondria. These observations were supported by Kuwana et al. (57), who reported that CL is required for Bid-Bax–mediated pore formation in liposomes. In mammalian cells, Bax oligomerization, which is necessary for Bax-mediated pore formation, requires tBid and occurs after Bax translocation to mitochondria. In contrast, using cardiolipin-deficient yeast mitochondria, Iverson et al. (58) demonstrated that the mitochondrial association of exogenous, recombinant Bax, and resultant cytochrome c release, occurred independently of the CL content of the yeast mitochondrial membranes. It was apparent, however, that cytochrome c was bound more “loosely” to the CL-deficient IMMs as compared to its binding to control IMMs. These data support a two-step mechanism of cytochrome c release (51).
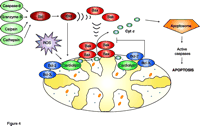
Regulation of cytochromecrelease from mitochondria. The mitochondrial outer membrane is permeabilized by tBid, which promotes the oligomerization and insertion of Bax and Bak. Cytochrome c release is initiated by the dissociation of the hemoprotein from its binding to cardiolipin in the inner mitochondrial membrane, which is stimulated by cardiolipin peroxidation (which is mediated by ROS). Once released into the cytosol, cytochrome c triggers pro-caspase-activation via the apoptosome mechanism.
The unsaturated nature of the acyl chains of CL renders this biomolecule susceptible to peroxidation by ROS, thus providing another mechanism by which CL levels may be reduced leading to apoptosis. The involvement of ROS in necrotic cell death has been known for decades, yet their involvement in apoptotic cell death was recognized relatively recently when it was reported that the anti-apoptotic proteins Bcl-2 and Bcl-XL protected cells against oxidants by shifting the cellular redox potential to a more reduced state (59). Cell toxicity induced by ROS occurs when the antioxidant capacity of the cell is overwhelmed and may be the consequence of oxidatively-modified DNA, lipid, or protein. Indeed, because ROS are “reactive” they will react with other molecules at, or near, the site of ROS production, with cardiolipin especially vulnerable because of its unsaturated carbon chains and proximity to ROS-producing sites in the IMM.
Receptor-mediated apoptosis induced by tumor necrosis factor –α (TNF α) provided the first evidence that mitochondrially generated ROS may be important in the execution of apoptosis by this cytokine (60). There is now widespread evidence that ROS are important mediators in apoptosis. Hence, the addition of ROS––or the depletion of endogenous antioxidants––can lead to CL peroxidation, cytochrome c release, and apoptosis in multiple experimental systems (50). These findings imply that ROS may not only cause direct toxicity, but that they also act as signals or mediators in the apoptotic process. In addition, overexpression of antioxidant enzymes, such as MnSod (61), PHGPx (62), and thioredoxin (63) confer protective effects on cells subjected to ROS-producing apoptotic stimuli. Recent studies of mitochondrial peroxiredoxin III (64) and glutaredoxin 2 (65) have further emphasized the protection from cardiolipin peroxidation, cytochrome c release and apoptosis offered by the mitochondrial antioxidant enzymes.
Most often, cytochrome c release occurs by permeabilization of the outer mitochondrial membrane by pro-apoptotic Bcl-2 family proteins, such as Bid, Bax, and Bak (48). Anti-apoptotic Bcl-2 proteins (e.g., Bcl-2 and Bcl-XL) inhibit pore formation by this mechanism (Figure 4⇑). Furthermore, mice deficient in both Bax and Bak are resistant to most apoptotic stimuli, thus providing strong support for the importance of this mechanism in the release of cytochrome c leading to the activation of caspases.
Before cytochrome c can be released into the cytoplasm via pore formation it must first become detached from CL, and there is mounting evidence that ROS might influence this aspect of cytochrome c release. For example, oxidative degradation of mitochondrial CL occurs during p53-induced apoptosis (66). As mentioned above, studies using bovine heart submitochondrial particles support the hypothesis that oxidative damage to CL is paralleled by mitochondrial dysfunction and cytochrome c release (53). Furthermore, a model of glutamate toxicity in neurons demonstrated that cytochrome c is released from mitochondria in a ROS-dependent fashion (67), and a burst of ROS in growth factor-deprived neurons was found to damage mitochondria by causing a profound loss of CL (68).
In conclusion, the two-step mechanism of cytochrome c release, connects the upstream event of cytochrome c (i.e., dissociation from CL) with its subsequent release through pores in the outer membrane (Figure 5⇓). Hence, cytochrome c must first dissociate from the inner membrane in order for it to escape the mitochondria. It was also demonstrated directly by HPLC–UV that CL becomes oxidized upon treatment with various oxidants, and that the amount of peroxidized CL correlates positively with cytochrome c release upon pore formation (51). Therefore, oxidation of CL may be one mechanism by which cytochrome c is solubilized in the intermembrane space, rendering it “available” for release into the cytosol upon permeabilization of the outer mitochondrial membrane. Other mechanisms might contribute to cytochrome c detachment under non-oxidant conditions, but they remain to be identified.
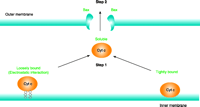
Cytochromecis released from mitochondria by a two-step mechanism. Pore formation by Bax is required, but not sufficient for release of the hemoprotein from the mitochondria. Cytochrome c must first be detached from its binding to cardiolipin in the IMM to be released into the cytosol through the pores in the outer membrane.
Role of Mitochondrial Aconitase in Oxidative Neurodegeneration
Mitochondrial oxidative stress has been implicated as a common feature in the etiology of diverse chronic neurodegenerative diseases, including Alzheimer Disease, Parkinson Disease (PD), Huntington Disease, and amyotrophic lateral sclerosis (69). Oxidative stress is generally defined as an imbalance that favors the production of ROS over antioxidant defenses; however, the precise mechanisms by which oxygen radicals produce cellular injury remain elusive. A central mediator that influences both physiological and pathological processes is O2·− (70), a by-product of mitochondrial respiration thought to comprise between 0.4–1% of total oxygen consumption (71). Because O2·− has limited reactivity, its ability to directly exert toxicity in pathophysiological processes has been questioned and led to the search for more potent oxidants, such as peroxynitrite (ONOO−) and hydroxyl radical (·OH), that may underlie O2·− toxicity. Experimental evidence over the past several decades, however, continues to support a role of O2·− in pathological processes. An important mechanism of O2·− toxicity is its direct oxidation and resultant inactivation of iron-sulfur (Fe-S) proteins, such as aconitases, and resultant release of iron (70). Although oxidative inactivation of Fe-S proteins is known to underlie O2·− toxicity in bacteria and yeast, whether this contributes to injury in the mammalian brain is unknown and is the focus of this section. The role of O2·− toxicity by its direct oxidation and consequent inactivation of aconitases is explored in experimentally induced PD.
The unique and preferential sensitivity of the reaction of O2·− with labile Fe-S proteins can be attributed to the presence of an unligated, acid-labile iron atom (Fea) in the active site of the enzyme [see (72) for review]. The electrophilicity of Fea in the Fe-S cluster of aconitase renders this (de)hydratase especially sensitive to oxidative inactivation by nucleophilic O2·−, involving the univalent oxidation of the [4Fe-4S]2+ cluster (73). Consequently, an unstable intermediate is formed, which fails to keep the unligated Fea bound to the cluster. This instability promotes the loss of Fea from the [4Fe-4S]2+ cluster—subsequently forming a [3Fe-4S]2+ cluster—with concomitant loss of enzyme activity. Not all Fe-S enzymes contain an unligated Fea motif; therefore, this sensitivity is not universal to all Fe-S centers. For example, Fe-S centers of xanthine oxidase and several of the mitochondrial electronic transport chain (ETC) complexes are not known to possess labile Fe-S centers that would render them susceptible to oxidative inactivation.
Whether O2·− mediates toxicity via oxidative damage to Fe-S enzymes depends on the abundance of the Fe-S enzymes, other competing targets for O2·− within its close vicinity, the rate of reaction of O2·− with these targets and the availability of substrates that may protect the Fe-S enzymes from inactivation. For example, the fate of mitochondrial O2·− is controlled by its reactivity with several important targets that include Sod2, nitric oxide (NO), and susceptible Fe-S centers. Based on estimated concentrations of Fe-S enzymes, of Sod2 and NO, and rates of reaction of O2·−with these targets, it has been estimated that in E. coli, 75% of O2·− reacts with labile Fe-S centers and 25% with NO (74). Therefore, labile Fe- S–containing enzymes such as aconitase, which are abundant in the brain, offer an important target for O2·−, particularly as steady-state [O2·−] increases and endogenous Sod2 is inactivated.
The inactivation of mitochondrial aconitase may have at least two major consequences. First, the formation of an inactive [3Fe-4S]+ cluster results in the simultaneous release of Fe2+ and H2O2. In fact, O2·− mediated inactivation of Fe-S-containing enzymes may pose a significant oxidative burden because it provides equimolar amounts of H2O2 per mole of O2·− (74). The release of Fe2+ and H2O2, ingredients of the Fenton reaction, can result in generation of the potent ·OH radical, which can oxidize mitochondrial proteins, DNA, and lipids thereby amplifying O2·− initiated oxidative damage. Aconitase inactivation is capable of generating iron and free radicals following posttranslational inactivation by O2·− has been verified experimentally. Keyer and Imlay first demonstrated the release of iron from E. coli aconitase and its involvement in O2·− mediated toxicity (75). Vasques-Vivar et al. (76) tested the idea that O2 − mediated inactivation of mitochondrial aconitase can be a source of ·OH formation by Fenton chemistry initiated by the co-released Fe2+ and H2O2. This study demonstrated the formation of ·OH, originating from mitochondrial aconitase upon oxidative inactivation by electron paramagnetic resonance, suggesting that purified aconitase is capable of generating ·OH in a cell-free system following oxidative stress. Whether aconitase inactivation results in free radical formation and toxicity in vivo remains unknown. Secondly, mitochondrial aconitase plays a key role in the TCA cycle, catalyzing the (reversible) dehydration of citrate to isocitrate. The dependence of ATP production via the ETC on the TCA cycle illustrates the impact any interruption in the TCA activity can have on cellular energy production. Even partial inhibition of mitochondrial aconitase could result in TCA dysfunction impacting energy production and, therefore, neuronal viability.
Based on the well-described role of Fe-S proteins in O2·− toxicity in bacteria and yeast, the hypothesis that oxidative inactivation of mitochondrial aconitase contributes to neuronal injury in the mammalian brain was tested. Mammalian cells contain two aconitases, cytosolic and mitochondrial forms, which are encoded by two different genes (77) both of which contain a labile [4Fe-4S] center in their respective active site that can catalyze the reversible isomerization of citrate to isocitrate via cis-aconitate. The active site residues are identical in both enzymes (78, 79). However, the cytosolic aconitase, also known as iron-responsive protein-1 (IRP1), is a bifunctional protein that binds to iron-responsive elements (IREs) and controls the amount of iron in cells (80, 81). IREs are RNA stem-loop sequences in the 5′ untranslated region (UTR) of the mRNAs for iron storage proteins (e.g., ferritin) and the 3′-UTR of iron-utilizing proteins such as the transferrin receptor. The IRE allows the IRPs (IRP1 and IRP2) to bind in response to changing iron levels and regulate iron utilization by controlling the synthesis or degradation of the IREcontaining protein. The majority of brain aconitase activity resides in mitochondria (82). Although, mitochondrial aconitase shares 25% sequence homology with its cytosolic counterpart, it is not known to function as an IRP (78). Instead, mitochondrial aconitase contains a conserved and functional IRE in the 5′-UTR of its mRNA (80), suggesting that IRPs could potentially regulate the levels of mitochondrial aconitase in response to changes in iron levels.
Mitochondrial oxidative stress, aging, iron imbalance, and environmental agents play important overlapping roles in PD; however, the precise mechanism(s) by which these factors mediate the death of dopaminergic neurons in PD remains unclear. Several common features between the etiological factors involved in pathogenesis of PD and inactivation of mitochondrial aconitase provide a compelling argument for examining the hypothesis that O2·− toxicity via mitochondrial aconitase inactivation contributes to neuronal injury in PD. First, among age-related neurodegenerative diseases, PD provides the best example of a disease in which a strong role for mitochondrial oxidative stress has been implicated (83). Because cumulative oxidative damage to cellular macromolecules is thought to underlie the process of aging, proteins such as mitochondrial aconitase may be preferential targets of age-related oxidative stress. In fact, mitochondrial aconitase is selectively oxidized and inactivated through the process of aging, in a fly model (84). In this study, a single protein, identified as mitochondrial aconitase was specifically oxidized as a function of aging in the fly flight muscle (84). Second, an important consequence of acute mitochondrial O2·− toxicity in sod2−/− mice is manifested by a dramatic inactivation of mitochondrial aconitase. More stable Fe-S containing enzymes are inactivated to a lesser extent (85). The sod2−/− mouse model underscores the importance of mitochondrial aconitase inactivation as a mechanism underlying acute mitochondrial O2·− toxicity. The sod2−/+ mice, however, are better suited for testing chronic mitochondrial O2·− toxicity that may mimic age-related pathological processes. The sod2−/+ mice are normal at birth and develop mitochondrial defects associated with increased O2·− production as they age (86), including inactivation of mitochondrial aconitase (87). Therefore, both acute and chronic mitochondrial O2·− toxicity is accompanied by inactivation of mitochondrial aconitase. Work in our laboratory demonstrates age-dependent inactivation of brain mitochondrial aconitase in sod2−/+ mice (16). Together, these observations suggest that the process of aging is a major risk factor common to oxidative stress, aconitase inactivation, and PD.
Finally, pathogenesis of PD is linked with an imbalance of iron. Several studies have shown iron accumulation by various methods including histochemical techniques in human and experimental PD and the protective role of iron chelators (88, 89). The sources and mechanisms of the iron imbalance in PD remain to be determined. Finally, because endogenous and environmental factors are thought to contribute to PD, mechanisms common to both causes may better explain the pathogenesis of the disease. Neurotoxins and environmental agents linked with the etiology of PD (90) (e.g., rotenone, paraquat, or MPTP) can inactivate mitochondrial aconitase in neuronal cultures (91), providing a rationale for determining the role of aconitase in animal models of PD.
A defect in mitochondrial ETC at Complex I is thought to account for increased steady-state mitochondrial O2·− formation in human and animal models of PD (92); however, whether the increased mitochondrial O2·− results in neuronal damage remains unclear.
The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model replicates some key features of PD (93), therefore this toxin-based model has been used to determine if mitochondrial aconitase inactivation and iron accumulation contribute to dopaminergic neuronal death. The involvement of O2·− radicals in MPTP neurotoxicity has been previously demonstrated using mutant mice overexpressing or deficient in Sod1 or Sod2 (94–97). MPTP-treatment inactivated O2·− sensitive mitochondrial aconitase in the vulnerable substantia nigra at times preceding overt neurotoxicity (16). Importantly, MPTP mobilizes a novel early pool of chelatable mitochondrial iron that parallels mitochondrial aconitase inactivation. Because Sod2 is both an efficient scavenger of O2·− and a modulator of mitochondrial aconitase activity, we utilized mice overexpressing or deficient in Sod2 to determine the role of mitochondrial O2·− in the mechanism of MPTP-induced aconitase inactivation and iron accumulation. MPTP-induced mitochondrial aconitase inactivation, chelatable iron, and striatal dopamine depletion are attenuated in mice that overexpress Sod2 and exacerbated in mice with a partial deficiency of Sod2, confirming the role of mitochondrial O2·− in these processes (16). Together, these observations suggest that O2 −mediated inactivation of mitochondrial aconitase and resultant iron release may contribute in part to dopaminergic neuronal death in the MPTP model of PD. These studies also reveal that chronic mitochondrial O2·− toxicity in sod2−/+ mice is manifested by an agedependent exacerbation of MPTP-induced mitochondrial aconitase inactivation, iron accumulation, and dopamine depletion.
As described in the preceding sections, inactivation of mitochondrial aconitase by increased O2·− is proposed to contribute, in part, to the death of dopaminergic neurons by the following mechanism (Figure 6⇓). O2·− mediated oxidation of the [4Fe-4S]2+- containing mitochondrial aconitase would result in a [3Fe-4S]+ inactive aconitase, Fe2+, and H2O2. The interaction of Fe2+ and H2O2 results in ·OH formation. Oxidative damage could ensue because of this increased free radical load and oxidant damage to mitochondrial proteins, lipids, and DNA. In addition to catalyzing the formation of ·OH, both Fe2+ and H2O2 released by aconitase inactivation can modulate gene expression and cell signaling, respectively. The short-term fate of the iron released from Fe-S clusters most likely involves association with low-molecular-weight chelators, such as citrate and ADP. Additionally, mitochondrial aconitase inactivation would result in TCA cycle inhibition and thereby worsen the already ongoing energy crisis. Furthermore, the increased free iron may bind and activate IREs in the mRNA of certain proteins (e.g., ferritin) and thereby modulate their translation (98). Such IRE-mediated regulation could result in a long-term change in iron homeostasis in the surviving tissue; however, the long-term consequences of this iron on cellular iron homeostasis remain to be determined. Such a mechanism could contribute to injury not only in PD, but a variety of conditions in which steady state levels of O2·− are elevated for prolonged time periods. For example, inactivation of aconitase has also been demonstrated in the brains of patients with Huntington Disease (99), in progressive supranuclear palsy (69), and in an animal model of status epilepticus (100). Based on the proposed mechanism, therapeutic intervention could be achieved with antioxidants, such as O2·− scavengers and iron chelators, particularly those agents that penetrate the mitochondrial compartment.
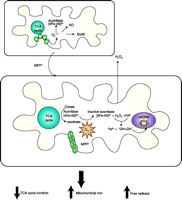
Proposed scheme of events in MPTP-induced mitochondrial O2·−toxicity via oxidative aconitase inactivation. The fate of mitochondrial O2·− is normally controlled by its reactivity with several targets that include Sod2, nitric oxide NO, and Fe-S centers. Elevated steady-state mitochondrial O2·− production resulting from Complex I inhibition by MPTP’s active metabolite, MPP+, would favor oxidative inactivation of the [4Fe-4S]2+-containing aconitase and simultaneous release of Fe2+ and H2O2. Subsequent generation of HO· via Fenton chemistry could damage mitochondrial proteins, DNA, and lipids thereby amplifying O2 −initiated oxidative damage. Additionally, the release of Fe may affect cellular iron homeostasis and decreased aconitase activity could interrupt its TCA cycle function. Adapted from (137).
In conclusion, mitochondrial oxidative stress is thought to be important pathological mediator of neuronal death in diverse neurodegenerative diseases, most notably PD. The precise mechanism by which mitochondrial oxidative stress injures vulnerable neuronal populations, however, remains unclear. An important premise of O2·− toxicity in lower organisms is based on its oxidative inactivation of critical Fe-S-cluster–containing proteins. Consequent release of redox-active iron in the mitochondrial milieu together with partial loss of TCA cycle function may serve as critical mechanisms responsible for neuronal injury. Based on its structure and location, mammalian mitochondrial aconitase is uniquely poised to participate in neurotoxic events resulting from elevated mitochondrial O2·− Examination of this idea in a neurotoxin-induced Parkinsonism suggests that mitochondrial aconitase inactivation and early iron accumulation may play a pathogenic role in PD.
Role of Mitochondrial Uncoupling Protein 2 in the Pathogenesis of Type 2 Diabetes
Uncoupling protein-2 (UCP2), which is expressed in several metabolically important tissues including pancreatic beta-cells, functions to uncouple OXPHOS in mitochondria, thereby reducing the cellular energy quotient (Figure 1⇑). Early in our understanding of the potential physiological and pathophysiological roles of UCP2, dual and opposing functions were hypothesized for tissues important to the development of type 2 diabetes, including white adipose, skeletal muscle, and endocrine pancreas (101). The common denominator linking UCP2 expression in the relevant tissues was free fatty acids (FFA), suggesting that in muscle and adipocytes, induction of UCP2 expression might be an adaptive response to elevated circulating FFA, which are found in obesity and type 2 diabetes, serving to limit the development of insulin resistance. In contrast, increased expression of UCP2 in response to FFA in pancreatic beta-cells appears to be detrimental to glucose-stimulated insulin secretion and may be an important contributor to diabetes development. Genetic analysis of the human UCP2 promoter region shows that the −866G/A polymorphism (A/A allele) associates with higher UCP2 expression, type 2 diabetes, and decreased insulin secretion although the degree of impact has varied from population to population (102–106). A common polymorphism in the coding region (55A/V) is linked with decreased energy expenditure (107) but not with insulin resistance (108), or diabetes (109). Although resting twentyfour hour energy expenditure is lower in V/V homozygotes, an increase in spontaneous physical activity normalizes the total energy expenditure (107). Even in the absence of polymorphisms that increase its expression, UCP2 expression and activity can be regulated by nutritional status and oxidative stress. This section will focus on the links between fatty acid metabolism, oxygen radical production, UCP2, and insulin secretion.
Insulin resistance, or inability of peripheral target tissues to take up glucose in response to insulin, is common to both obesity and type 2 diabetes. Yet, in simple obesity, glucose tolerance is relatively normal. In genetically susceptible individuals, prolonged mild hyperglycemia combined with elevated triglyceride and FFA in plasma causes a condition called “glucolipotoxicity” that leads to impaired glucose-stimulated insulin secretion to the extent that hyperglycemia eventually exceeds the threshold for diagnosis of diabetes. In isolated islets, exposure to FFA in the presence of minimally stimulatory glucose concentrations for forty-eight hours mimics glucolipotoxicity. An increase in basal insulin secretion is observed, but the beta cells fail to respond to incremental increases in glucose (110, 111). Although multiple changes in beta-cell function occur in glucolipotoxicity (112), UCP2 may be a central regulator, contributing to the control of coupled oxidative metabolism, cellular energy production, and response to oxygen radical production.
Mitochondrial metabolism, particularly the degree of substratestimulated hyperpolarization of the IMM, determines the ability of the beta cell to respond to stimuli (113). Thus, an increase in uncoupled respiration would be predicted to suppress insulin secretion. Several key findings support the hypothesis that UCP2 negatively regulates insulin secretion and may be involved in the pathogenesis of type 2 diabetes. First, overexpression of UCP2 suppresses glucose-stimulated insulin secretion (114–116) whereas ucp2−/− mice exhibit elevated insulin responsiveness to glucose both in vivo and in vitro (117). Second, in most animal models of obesity and diabetes studied, UCP2 expression in beta cells is increased (117–119). Third, feeding normal mice a high fat diet concomitantly induces a diabetic beta-cell phenotype and increased UCP2 expression, whereas beta cells from mice lacking UCP2 maintain high glucose responsiveness (111, 120). The expression of UCP2 is transcriptionally regulated by peroxisome proliferator-activated receptors (PPARs) (121, 122) and by sterol response element binding protein-1c (SREBP-1c) (123). Exposure of islets to elevated glucose also increases transcription of ucp2 (124). The simplest explanation, substantiated by measurement of mitochondrial membrane potential and cellular ATP concentrations (111, 115), is that increased uncoupling of OXPHOS after induction of ucp2 expression impairs the ability of beta cells to generate ATP in response to glucose. Because ATP is a source of high-energy phosphate bonds and is also an important signaling molecule in beta cells, a decrease in ATP production has multiple consequences for insulin secretion [see (125) for review]. The link between glucolipotoxicity and UCP2 involves, however, more than transcriptional regulation of expression (Figure 7⇓).
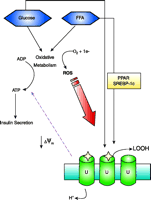
Schematic representation of the hypothesized relationship between glucolipotoxicity, ROS, and UCP2 (U) in pancreatic beta-cells. Exposure of beta cells to glucose and FFA increases expression of UCP2. Both peroxisome proliferator-activated receptors (PPAR) and sterol response element binding protein-1c (SREBP-1c) have been implicated in induction of UCP2 transcription. Oxidative metabolism of glucose and FFA generates ATP. ROS such as superoxide are a by-product of oxidative phosphorylation, with production increasing under conditions of chronic over-nutrition. ROS-mediated activation of UCP2 (stars) lowers the mitochondrial membrane potential (ΔΨm), resulting in a decrease in ATP production to attenuate glucose-stimulated insulin secretion. Although ultimately detrimental, the activation of UCP2 is an attempt to protect beta cells from further ROS production and to export harmful lipid peroxides (LOOH) from the mitochondria.
Based on studies in beta-cell lines, in which application of exogenous H2O2 induces UCP2 expression and inhibits insulin secretion, the potential for oxygen radicals to be involved in UCP2’s mechanism of action was revealed (126). Moreover, overexpression of UCP2 in the beta-cell line was protective against H2O2-induced cell death (126). FFA can induce uncoupling of oxidative phosphorylation, possibly via UCP2 (127), and exposure of beta cells to the FFA oleic acid (OA) reduces the ATP:ADP ratio and depolarizes the mitochondrial membrane coincident with induced expression of UCP2 (110). Several groups have found that the production of ROS is stimulated by OA in beta-cell lines (128, 129) and by palmitic acid (PA) in rat islets (111, 127) although others did not (130). Whether FFA-stimulated ROS directly stimulate UCP2 expression has not yet been established; in fact, two studies showed that beta cells cultured with FFA and antioxidants were not protected from FFA-mediated inhibition of insulin secretion (130, 131).
To determine the potential link between obesity or high fat diets with increased UCP2 activity, insulin-secreting MIN6 cells were exposed to OA or PA for forty-eight hours. This treatment led to increased sensitivity to the subsequent acute uncoupling action of FFA, an effect shown to be mediated by O2·−. The latter point was proven by preincubating cells with cell-permeant O2·− scavengers, which blocked the effects of OA (132). Moreover, OA-exposed cells produced five-fold more O2·− than control cells. Thus, although it is not yet established that ROS mediate the FFA-related increase in UCP2 expression, FFA does potentiate ROS production in the mitochondria, leading to increased uncoupling and dissipation of the mitochondrial membrane potential (132).
Beta cells exposed to high concentrations of glucose also generate more ROS (133), and exposure of mouse islets to 25 mM concentrations of glucose for seventy-two hours leads to increased UCP2 expression (124). In the absence of UCP2, hyperglycemic culture conditions increase mitochondrial ROS production more than that observed in islets expressing UCP2; however, superoxide fails to inhibit glucose-stimulated insulin secretion (124). These authors concluded that the inhibition of insulin secretion by O2·− was mediated by UCP2, consistent with data that O2·− activates UCP2 in isolated mitochondria (134). Moreover, when ob/ob mice were crossed with ucp2−/− mice (ob/ob/ucp2−/−), the insulin responsiveness of ob/ ob/ucp2−/− islets to glucose was enhanced relative to responsiveness observed in native ob/ob islets (117, 124). These effects were mimicked by treating ob/ob islets with antioxidant enzymes (124).
In summary, data from studies presented here are largely consistent with the idea that at least one consequence of glucolipotoxicity is increased expression and activity of UCP2 in beta cells, leading to impaired glucose-stimulated insulin secretion. A schematic representation of the hypothesized relationship between glucose–FFA, ROS, and UCP2 is presented in Figure 7⇑. Generation of O2·− or other ROS by either FFA or glucose metabolism is a key component of UCP2 activation. It was recently proposed that O2·− exerts its activating effects on UCPs indirectly by catalyzing the generation of lipid peroxides and their breakdown products, reactive alkenals (135). Additionally, UCPs might participate as carriers of lipid peroxide anions to protect cells against oxidant damage (136). Moreover, outward movement of anions will dissipate the mitochondrial membrane potential gradient, which would help prevent further accumulation of O2·−. Unfortunately, in the beta cell, such protection may come at a cost of impaired insulin secretion to the extent that type 2 diabetes ensues.
Conclusion
Considerable evidence provided in this review supports the importance of mitochondria and mitochondrial oxidative damage, respectively, as a critical target and event responsible for toxic oxidative stress and related diseases. This is supported, in part, by the demonstration of the dramatic protection of cells against toxic oxidative stress following the enrichment of mitochondrial membranes with vitamin E. The specific mitochondrial target(s) oxidatively damaged that are directly responsible for cell injury and disease, are more difficult to define; however this review provides compelling information to support the importance of four different mitochondrial targets in toxic oxidative stress. Evidence was provided in this review for the importance of mtDNA integrity in the maintenance of cell viability as well as for the importance of the oxidation of IMM-bound cardiolipin as a critical event in the mitochondrial release of cytochome c and the initiation of apoptosis. Furthermore, oxidative stress-mediated inhibition of mitochondrial aconitase with the concomitant release of bioavailable mitochondrial iron appears to play a critical role in neuronal cell death related to PD. Finally, evidence was offered that provided a role for mitochondrial uncoupling protein 2––induced by hyperglycemia- and fatty free acid-mediated oxidative stress—in preventing mitochondrial ROS generation in beta cells but also limiting ATP production and, thus, glucose-stimulated insulin secretion (involved in type 2 diabetes pathogenesis). Undoubtedly, future mechanistic studies on the role of mitochondria in toxic oxidative stress will provide us with a better understanding of the critical mitochondrial events that are responsible for oxidative stress–mediated diseases as well as provide us with cellular targets to develop effective therapeutic agents for numerous common diseases.
Acknowledgments
This review represents a concise overview of the authors’ scheduled presentations at the ASPET, Division of Toxicology Symposium entitled “Role of Mitochondria in Toxic Oxidative Stress” during the 2005 Experimental Biology meeting in San Diego, CA. The work in the authors’ laboratories were supported as stated below for each author: MWF by grants from the National Institute of Health (NS044202, ES05452) and the Lazzara Foundation. Work presented in this review on mtDNA damage and repair was performed by Astrid Haugen, and Drs. Janine H. Santos and Michael Yakes, to whom BVH is indebted for their outstanding efforts. MP by a grant from the National Institute of Health (NS045748). CBC by grants from the Canadian Institutes of Health Research and the Canadian Diabetes Association.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Sten Orrenius, MD, PhD, is a Professor in the Divison of Toxicology, Institute of Environmental Medicine at the Karolinska Institute, Stockholm, Sweden. His research interests center on investigating the regulation of toxic cell death. Please address E-mail correspondence to SO at sten.orrenius{at}imm.ki.se

Bennett Van Houten, PhD, is a Principal investigator at the National Institute of Environmental Health Sciences (NIEHS), in the Laboratory of Molecular Genetics, Research Triangle Park, NC, 27709. His laboratory is interested in structure- function studies of DNA repair enzymes, and the role of mtDNA damage in cell death. Please address E-mail correspondence to BVH at vanhout1{at}niehs.nih.gov

Manisha Patel, PhD, is an Assistant Professor in the Department of Pharmaceutical Sciences at the University of Colorado Health Sciences Center, Denver. Her research interests include understanding mechanisms of oxidant production in neurological disorders, identifying oxidant-sensitive cellular targets and developing neuroprotective antioxidant therapies. Please address E-mail correspondence to MP at manisha. patel{at}uchsc.edu

Cathy Chan, PhD, is a Professor of Physiology in the Department of Biomedical Sciences at the Atlantic Veterinary College, University of Prince Edward Island, Canada. Her research interests include characterization of the functional changes in beta-cell function in obesity and type 2 diabetes, with a current emphasis on the role of uncoupling protein-2 in development of lipotoxicity in islets. Please address E-mail correspondence to CBC at cchan{at}upei.ca

Marc W. Fariss, PhD, is an Associate Professor of Toxicology at the University of Colorado Health Sciences Center and Cancer Center. His research interests include defining the protective role of mitochondrial vitamin E in oxidative stress-mediated diseases such as iron overload liver disease, diabetic nephropathy, Parkinson Disease and cancer. Address E-mail correspondence to MWF at marc.fariss{at}uchsc.edu; fax 303-315-0274.

