TLRs: Differential Adapter Utilization by Toll-Like Receptors Mediates TLR-Specific Patterns of Gene Expression
Abstract
In response to microbial or environmental “danger” signals, represented by structural motifs not normally expressed by cells, Toll-like receptors mediate intracellular signaling that leads to inflammatory gene expression. In response to agonists, TLR aggregation enables the recruitment and/or activation of TLR-specific adapter molecules. To date, four adapter proteins have been identified: MyD88, TIRAP/Mal, TRIF/TICAM-1, and TIRP/TRAM/TICAM-2. The interaction of the different TLRs with distinct combinations of adapter molecules creates a platform to which additional kinases, transacting factors, and possibly other molecules are recruited, events that lead, ultimately, to gene expression. Given the rapidity with which such interactions have been described, we have attempted to summarize our current understanding of the adapters that are so essential for TLR signaling and provide a working model for future studies.
Introduction
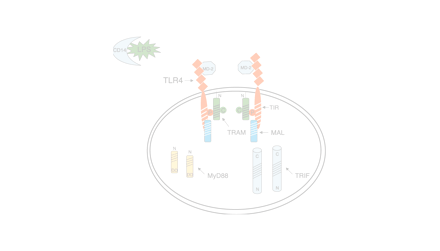
In response to agonist-induced activation through the extracellular N terminus, Toll-like receptors (TLRs), which belong to the type I family of membrane-spanning receptors, aggregate as homo- or heterodimers, and also form larger aggregates (1). Transduction of intracellular signaling depends upon the constitutive association and/or recruitment of specific adapter molecules to the intracytoplasmic C terminus through structurally conserved motifs called “Toll–Interleukin (IL)-1 Resistance” (TIR) domains present in both TLRs and adapter molecules (Figure 1A⇓). The adapters, in turn, provide a structural platform to which various kinases and substrates are recruited. A cascade of biochemical reactions is initiated at the platform and these culminate in the activation of pathway-specific DNA binding proteins that initiate transcription. In other words, the specific combination of adapter molecules that associate with a particular TLR ultimately dictates the distinct pattern of gene expression that one observes in response to different TLR agonists. Hirschfeld et al. (2) provided the first evidence of differential gene expression induced by TLR4 vs TLR2 activation in macrophages by comparing expression of a limited panel of pro-inflammatory genes. These findings have since been confirmed and extend to several TLRs, most recently, by employing DNA microarrays that permit a much more extensive analysis of the gene-inducing capabilities of different TLRs (3–5). Four adapters are known to mediate TLR signaling and share significant amino-acid sequence similarity within their TIR domains (6), including (in order of their discovery): myeloid differentiation factor 88 (MyD88); TIR domain-containing adapter protein (TIRAP), also called, MyD88 adapter-like (Mal); TIR domain-containing adapter inducing interferon (IFN)-β (TRIF), also called TIR domain–containing adapter molecule-1 (TICAM-1); and most recently, TIR-containing protein (TIRP), also referred to as TRIF-related adapter molecule (TRAM) or TICAM-2. Aligned amino acid sequences of the TIR domains for the four known TLR adapters are depicted in Figure 1B⇓. A fifth TIR domain-containing protein called SARM (for “Sterile alpha and HEAT/Armadillo motif protein”), which encodes an ortholog of Drosophila and C. elegans proteins, also exists in mammals. Although SARM also contains a TIR domain in its C terminus (Figure 1A⇓), its role in TLR signaling is currently unclear (6). It is our view that upon agonist-induced stimulation the unique association of specific adapters with each TLR enables a specific signaling program that ultimately leads to a TLR-specific repertoire of gene expression.

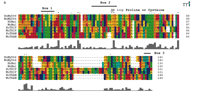
Schematic representation of known mammalian TLR adapter molecules. A. MyD88, TIRAP/Mal, TRIF/TICAM-1, and TIRP/TRAM/TICAM-2 are all known TLR adapter proteins, while the function of SARM is not yet known. B. CLUSTAL W alignment of the TIR domains of human and mouse MyD88, TIRAP/Mal, TRIF/TICAM-1, and TIRP/TRAM/TICAM-2. The amino acid colors are based on their physicochemical properties where yellow = small, green = hydrophobic, turquoise = aromatic, blue = positively charged and red = negatively charged. The three important regions, Box 1, 2, and 3 are labeled.
Different Methodologies Lead to Different Conclusions on Adapter Function
Limitations in available approaches for dissecting intracellular signaling pathways suggest that, no matter how thorough the study, only an incomplete picture can be discerned from any given study. In particular, a major problem in sorting through the extensive TLR adapter literature is that the approach used most frequently to evaluate the role of a specific adapter depends upon agonist-independent overexpression to drive signaling or to enable detection of intermolecular interactions by co-immunoprecipitation. However, overexpression of one or more signaling components disrupts the physiologic stoichiometry and may favor, or even create, protein–protein interactions through TIR domains that might not occur under normal circumstances of agonist-driven stimulation. Furthermore, it is often not clear if these interactions occur in all of the cells transfected or only in those that express the highest levels of proteins.
Overexpression of dominant-negative (DN) mutants is frequently used to demonstrate participation of a particular adapter molecule in TLR signaling. Although some DN mutants encode truncated forms of the adapter, many have been constructed such that a conserved proline (or cysteine, as in the case of TIRP/TRAM/TICAM-2; see below) within Box 2 of the BB loop of the TIR domain (Figure 1B⇑) is mutated to histidine (i.e., P/H or C/H). When overexpressed, such mutant proteins might not preclude interaction of the wild type, endogenous adapter through its BB loop with its target protein, but rather, would be predicted to sequester other adapters or other essential signaling molecules (e.g., kinases, or transacting factors) through interactions not involving the BB loop (7).
A less frequently employed approach to determine TLR adapter function is to use cell-permeable peptides that contain a Drosophila antennapedia sequence that enables the peptide to enter the cell, coupled to the ~15 amino acids that encompass and flank the conserved proline of the adapter’s BB loop. Although the mechanism by which such peptides inhibit signaling has not been formally demonstrated, it is likely that these small peptides inhibit BB loop–dependent interaction of the endogenous adapter with its natural target, possibly disrupting assembly of a larger signaling complex (7).
A relatively recent approach that has been applied to decipher the role of the adapter molecules in TLR signaling involves the use of small interfering RNA (siRNA). siRNAs function by directing the sequence-specific degradation of mRNAs that contain the siRNA sequence (8). Degradation of the target mRNA leads to reduction in the level of the cognate protein, and ultimately, to a detectable phenotype. This technique represents an exciting, valuable, and enabling tool to “knock down” gene function in mammalian cells. This approach has been used successfully to elucidate the role of adapters in TLR signaling and, in some cases, preceded the generation of adapter-deficient knockout mice that revealed identical phenotypes.
Experiments using mice that harbor targeted mutations within adapter genes have provided a wealth of information, but must also be viewed with caution. For instance, compensatory signaling mechanism(s) in knockout (KO) mice may overcome the lack of an adapter; the manner in which the gene was targeted may result in a mutant protein that exerts DN activity; or the lack of backcrossing the KO mice may result in a phenotype that is mistakenly attributed to the missing adapter.
Other technical considerations may underlie differences ascribed to the actions of various adapters in TLR signaling. It is often assumed that immunoprecipitation reflects direct contact between proteins from cell lysates; however, a bridging molecule that goes undetected by immunoblot analysis may mediate the apparent interaction. Alternatively, different proteins might be colocalized, but not physically associated, in membrane regions not disrupted by typical detergent solubilization methods (e.g., lipid rafts). It is also possible that direct protein–protein interactions, as measured in yeast two-hybrid systems, may be missed if either the “bait” or “prey” requires chemical modification (e.g., phosphorylation at the site of the receptor) for the interaction.
More subtle concerns stem from the fact that expansion of bone marrow-derived progenitor cells in Colony Stimulating Factor (CSF)-1 or granulocyte–macrophage CSF (GM-CSF) is a common approach to obtain macrophages and dendritic cells (DC), respectively, for in vitro analysis. However, CSF-1 is a potent inducer of interferon (IFN)-β (9), a cytokine whose expression is not induced by all TLRs. Therefore, if the “readout” for an experiment is the expression of IFN-β or IFN-β -dependent genes, the findings might well be confounded by the presence of CSF-1–dependent signals that are independent of the TLR pathway being studied, and might possibly lead to “priming” for a more robust response to LPS challenge (10). Lysates of L929 cells, often used as a source of CSF-1, may contain amounts of IFN-α /β sufficient to induce TLR-independent macrophage differentiation (11).
Adapters in the TLR Pathway
Despite the various caveats discussed above, extraordinary studies have been undertaken on the adapters that mediate TLR signaling. We will first review the roles of MyD88 and TIRAP/Mal in TLR signaling, and then focus on the functional interplay between these and two recently described adapters, TRIF/TICAM-1 and TIRP/TRAM/TICAM-2, which have been implicated in the “MyD88-independent” pathway.
MyD88
MyD88 is recruited to the C-terminal TIR domain of the IL-1 receptor (IL-1R), and mediates downstream signaling through recruitment of IL-1R–associated kinase (IRAK) to its N-terminal “death domain” (DD) (12, 13). Overexpression of a MyD88 mutant construct that lacks the C-terminal TIR domain induces NF-κ B activation, but does not interact with TLR4, suggesting the importance of MyD88 in TLR signaling (14). The MyD88 TIR domain alone acts as a DN inhibitor of TLR4-mediated NF-κ B activation (14).
The molecular basis for the unresponsiveness of the C3H/HeJ mouse strain to Gram-negative lipopolysaccharide (LPS) has been identified as a point mutation (P712H) within the BB loop of TLR4 (15, 16). Xu et al. (17) reported that the analogous mutation in TLR2 (P681H) precludes the interaction of TLR2 with MyD88, whereas overexpression of this same mutant construct inhibits TLR2-mediated signaling (18). Therefore, the C3H/HeJ signaling defect was presumed to be secondary to the failure of the P712H TLR4 to bind MyD88. Consistent with this hypothesis, Rhee and Hwang (19) showed that overexpression of a constitutively active TLR4 mutant recruits MyD88, but not if the construct expresses the P712H mutation. Overexpression of a MyD88 P200H mutant construct activates NF-κ B (20), indicating that this proline in the BB loop of MyD88 is not necessary for initiating downstream signaling. In contrast to Rhee and Hwang, however, Dunne et al. (21) found that overexpression of a human TLR4 (hTLR4) P712H mutant construct binds to MyD88 and conversely, that MyD88 (P200H) binds wild-type hTLR4. Thus, the conserved proline residue in MyD88 may not be required for it to interact with the TLR4 TIR domain, whereas the necessity of the TLR4 conserved proline for recruitment of MyD88 remains controversial.
The importance of MyD88 in LPS-mediated TLR4 signaling in vivo was first demonstrated by Kawai et al. (22). MyD88 KO mice were insensitive to LPS-induced death and failed to secrete cytokines such as IL-6 and TNF-α in vivo, or in vitro in LPS-stimulated macrophage cultures. Kawai et al. also observed this deficiency at the level of IL-6, TNF-α, and IL-1β mRNA expression. Although decreased amounts of TNF-α and IL-1β secretion and IL-6 mRNA have been confirmed by some, we and others have been unable to reproduce these findings for TNF-α, IL-1β, IFN-β mRNA and other LPS-inducible genes, although LPS-inducible IL-6 mRNA expression is depressed in MyD88 KO macrophages (23, 24, and S. Vogel and M. Fenton, unpublished observations). The basis for this discrepancy is presently unknown, but raises the possibility that MyD88 regulates post-transcriptional events that lead to cytokine secretion or, possibly, that differences in genetic backgrounds of the KO mice alter the phenotype at the level of mRNA. Most importantly, this seminal study by Kawai et al. revealed that key signaling pathways leading to LPS-induced NF-κ B translocation and phosphorylation of mitogen-activated protein kinases (MAPK) were slightly delayed, rather than eliminated, indicating the existence of a MyD88-independent pathway of TLR4 signaling. A recent study indicates that 76% of 913 genes induced in LPS-treated wild-type macrophages were induced in LPS-treated MyD88 KO macrophages (Björkbacka et al., unpublished results). In an earlier study, bone marrow-derived MyD88 KO macrophages and DCs stimulated with polyI:C (a TLR3 agonist) were profoundly impaired in their capacity to secrete nitric oxide or IL-12, respectively, under conditions where Iκ Bα degradation and MAPK phosphorylation in macrophages, and DC maturation, were normal (25). This suggested that TLR3-mediated signaling may also bifurcate into MyD88-dependent and independent pathways, an idea directly supported by the recent demonstration that treatment of MyD88 KO and wild-type macrophages with either LPS or polyI:C led to comparable induction of RANTES (r egulated upon a ctivation, n ormal T -cell e xpressed and s ecreted) secretion and activation of IRF-3, a transcription factor necessary for the expression of IFN-β and RANTES genes (26). The role of MyD88 in TLR3 signaling, however, remains controversial and will be discussed below.
TIRAP/Mal
Like MyD88, the second TLR adapter to be identified––termed TIRAP by Horng et al. (20) and Mal by Fitzgerald et al. (27) ––possesses a C-terminal TIR domain, but lacks the N-terminal DD. Overexpression of TIRAP/Mal activates NF-κ B, but this activation is blocked by the overexpression of MyD88 TIR or by the addition of inhibitors of other downstream signaling proteins (e.g., Iκ B super-repressor for NF-κ B or JNK-interacting protein for JNK) or other DN constructs (e.g., IRAK-2, TRAF-6, and TAK-1). However, overexpression of the TIRAP/Mal P125H mutant alone could not activate NF-κ B. Overexpression of the TIRAP/Mal TIR or P125H mutants blocked TLR4 signaling, but not signaling through IL-1R, IL-18R, TNFR, or TLR9. TIRAP/Mal appears to homodimerize and to associate constitutively with TLR4, but not with TLR9. Both TIRAP/Mal (20, 27) and TIRAP/Mal P125H (K. Fitzgerald, unpublished observation) interact with MyD88. A cell-permeable TIRAP/Mal blocking peptide (20) inhibited LPS-, but not CpG- (TLR9)-induced IL-12 and IL-6 expression, or antigen presentation by DC, indicating that TLR9 signaling is TIRAP/Mal-independent.
Dunne et al. (21) recently modeled TIR domain interactions based on electrostatic surface representations and hypothesized that TIRAP/Mal and MyD88 bind to different regions of TLR2 and TLR4, and interact with each other at a distinct non-overlapping site. In support of this model, these investigators were unable to provide any evidence that the conserved proline in the BB loops of TLR4, MyD88, or TIRAP/Mal contributes to any interaction amongst these molecules. Their findings are inconsistent with those of Horng et al. who found that TIRAP/Mal P125H and TLR4 did not associate, and with those of Rhee and Hwang who found that a TLR4 P712H mutant did not interact with MyD88. Direct interaction between TIRAP/Mal and TLR4 was demonstrated by glutathione S -transferase (GST)–TIRAP/Mal interaction with in vitro translated TLR4 (20), whereas yeast two-hybrid analyses demonstrated that MyD88 and TIRAP/Mal both homodimerized, and interacted with each other (27). MyD88 also interacted with TIRAP/Mal P125H mutant, supporting the notion that the TIRAP/Mal proline is not involved in its binding to MyD88.
Toshchakov et al. (24) showed, in murine macrophages, that TLR4- but not TLR2-mediated signaling induces the expression of IFN-β that, in turn, acts in an autocrine manner through IFN-α /β receptors to mediate the tyrosine phosphorylation of signal transducer and activator of transcription 1 (STAT1) and induction of STAT1-dependent genes. These authors found that LPS (TLR4)-induced, but not CpG DNA (TLR9)-induced, IFN-β gene expression was blocked by a TIRAP/Mal P125H DN construct and a cell-permeable inhibitory peptide composed of a Drosophila antennapedia sequence and the fourteen amino acids that include and flank the critical proline (i.e., P125) within the BB loop of TIRAP/Mal (20), whereas induction of IFN-β mRNA and increased STAT1 phosphorylation were normal in MyD88 KO macrophages. These findings supported a model in which TLR4 and TLR2 share a MyD88-dependent signaling pathway, whereas only TLR4 utilized TIRAP/Mal to activate a MyD88-independent pathway to induce IFN-β and STAT1-dependent genes that were not induced by TLR2 agonists (28). The TIRAP/Mal DN mutant also inhibited IRF-3 activation when induced by LPS, but not by polyI:C (TLR3) (29 ; K.A. Fitzgerald, unpublished), supporting the existence of distinct TLR signaling pathways that result in the induction of IFN-β expression.
With the advent of TIRAP/Mal KO mice (30, 31), it became clear that this model of distinct TLR signaling would have to be modified: LPS-induced cytokine production is severely impaired in TIRAP/Mal KO macrophages; however, responses to flagellin (TLR5), the immune response modifier resiquimod (R-848) (TLR7), CpG DNA (TLR9), IL-1, and IL-18 were normal in these macrophages. Unexpectedly, TLR2 signaling in TIRAP/Mal KO splenocytes and macrophages was also mitigated. Like MyD88 KO macrophages, TIRAP/Mal KO macrophages showed delayed NF-κ B translocation and MAPK phosphorylation induced by LPS, whereas TLR2-mediated NF-κ B translocation was essentially eliminated. The kinase activity of IRAK-1, stimulated by TLR2 or TLR4, was also lost in TIRAP/Mal KO macrophages.
Bone marrow-derived DC from TIRAP/Mal–MyD88 double KO mice respond normally to LPS to increase expression of cell-surface maturation markers, providing more support for separate TLR4-originating signaling pathways because TIRAP/Mal KO DC fail to secrete cytokines in response to LPS (TLR4) or bacterial lipopeptide (TLR2). LPS-induced activation of IRF-3 is normal in TIRAP/Mal KO macrophages (30), in contrast with earlier observations that the TIRAP/Mal DN mutant can block IRF-3 activation and LPS-induced IFN-β gene expression (24, 29). Nonetheless, data culled from TIRAP/Mal KO mice clearly indicate that TIRAP/Mal preferentially contributes to TLR2- and to TLR4-mediated signaling. TLR2 can form heterodimers with TLR1 or TLR6; thus, TIRAP/Mal might bind to TLR1 or TLR6 to facilitate the association. TIRAP/Mal associates with both TLR2 and TLR1, as demonstrated by pairwise transfections in HEK 293 cells, followed by co-immunoprecipitation (K. Fitzgerald, unpublished); however, the ability of TIRAP/Mal to interact with TLR6 has not been tested.
TRIF/TICAM-1
A third TIR-containing TLR adapter, called TRIF or TICAM-1, contributes to TLR3- and to TLR4-mediated signaling (32–35). TRIF/TICAM-1 is a much larger protein than either MyD88 or TIRAP/Mal—712 amino acids vs 296 and 235, respectively. Overexpressed TRIF/TICAM-1 also results in NF-κ B activation; however, only overexpressed TRIF/TICAM-1, but not MyD88 or TIRAP/Mal, leads to strong expression of an IFN-β gene reporter (32, 33).
Conversely, overexpression of MyD88 DN or TIRAP/Mal P125H DN fails to block polyI:C (TLR3)-induced activation of NF-κ B and IFN-β promoter activities, while these same DN constructs inhibit the relatively strong NF-κ B activity induced by the synthetic lipopeptide MALP-2, a TLR2 ligand. In contrast, overexpression of the TRIF/TICAM-1 TIR domain blocks both NF-κ B and IFN-β gene reporter activities induced by polyI:C (TLR3) (32), suggesting that MyD88 is not required for signaling [in contrast to data obtained by Alexopoulou et al. (25) using MyD88 KO mice]. In light of the recent controversy regarding the contribution of MyD88 to TLR3 signaling, we have carefully examined this issue. In MyD88-deficient macrophages or MyD88-deficient dendritic cells, polyI:C-mediated induction of TNF-α, IL-6, IP-10, and RANTES was comparable to that seen in wild-type cells (K. Fitzgerald et al., unpublished data). Importantly, the polyI:C used in these studies was free of contaminating endotoxin in contrast to other commercial sources. Therefore, preparations of polyI:C used in previous studies may have contained low levels of contaminating LPS that contributed to MyD88-dependent signaling through TLR4. These data, therefore, support the notion that TLR3 is unique in that it does not require MyD88 to mediate downstream signaling pathways.
Yamamoto et al. (32) and Oshiumi et al. (33) engineered TRIF/TICAM-1 deletion mutants, but their results are difficult to compare directly. The data of Yamamoto et al. suggest that a mutant construct that contains the N terminus plus TIR domain elicits nearly the same level of activation as wild-type TRIF/TICAM-1, but that neither the TIR domain alone nor the TIR domain plus the C terminus were active (32). Yamamoto et al. also reported that the TRIF/TICAM-1 TIR domain also exerts DN activity on TLR2-, TLR4-, TLR7-, MyD88- and TIRAP/Mal-driven NF-κ B expression, in addition to its capacity to inhibit TLR3-mediated IFN-β reporter activity (32). This suggests that the TRIF/TICAM-1 TIR DN construct is either not specific for TLR3 or that TRIF/TICAM-1 plays an as yet unappreciated role in these diverse TLR signaling pathways. Unfortunately, these authors did not test the TRIF/TICAM-1 TIR DN mutant for its ability to block TLR4- or TLR9-induced IFN-β reporter activity. More recently, however, Fitzgerald et al. found that the TRIF/TICAM-1 TIR DN mutant blocks TLR4 signaling to a RANTES promoter-driven reporter gene (26) and NF-κ B (K. Fitzgerald, unpublished); however, the DN mutant is a much stronger inhibitor of TLR3- than TLR4-mediated signaling. TLR3-stimulated IFN-β reporter activity was also inhibited by the TRIF/TICAM-1 TIR P434H DN (26).
In contrast to other TLRs, TLR3 has an alanine residue, A795, rather than the conserved proline in the BB loop of its TIR domain. Wild-type TLR3, but not the TLR3 A795H mutant, interacts with TRIF/TICAM-1 in an agonist-independent manner; however, Oshiumi et al. (33) found that TLR2 and TLR4 failed to associate with TRIF/TICAM-1, whereas Yamamoto et al. (32) showed that TLR2 could be immunoprecipitated with TRIF/TICAM-1. Yamamoto et al. also provided evidence for an interaction between TRIF/TICAM-1 and IRF-3––findings that have since been confirmed elsewhere (36). Overexpression of TRIF/TICAM-1 in HeLa cells resulted in IRF-3 activation, consistent with its ability to drive IFN-β reporter activity, and the use of small inhibitory RNA (siRNA) to decrease expression of TRIF/TICAM-1 or TLR3 resulted in ~50% inhibition of IFN-β secretion induced by TLR3 activation (33). Together, these data support a role for TRIF/TICAM-1 in the activation of IFN-β gene expression as a consequence of IRF-3 activation. The potent capacity of the TRIF/TICAM-1 TIR DN to inhibit NF-κ B driven by overexpression of other TLRs, despite the fact that they apparently do not interact with this adapter, raises concerns about the specificity of this DN: perhaps the TRIF/TICAM-1 TIR can bind and sequester TIRAP/Mal or MyD88.
TRIF/TICAM-1 KO mice express a similar phenotype to TLR3 KO mice: their macrophages are extremely deficient in their capacity to induce IFN-β, RANTES, IP-10, monocyte chemoattractant protein-1 (MCP-1) gene expression, and their B cells can neither proliferate nor increase the expression of activation markers in response to TLR3 (34). The lack of TRIF/TICAM-1 also affects LPS-induced cytokine gene expression in macrophage cultures, but does not alter IL-12 p40 induced by peptidoglycan (via TLR2), R-848 (via TLR7), or CpG DNA (via TLR9). Even in the presence of IFN-β, LPS did not stimulate production of detectable TNF-α, IL-6, or IL-12 p40 by TRIF/TICAM-1 KO macrophages, and B cell activation was also impaired. Not surprisingly, TRIF/TICAM-1 KO fibroblasts and macrophages failed to activate IRF-3. NF-κ B is activated by LPS in these cells, but not by polyI:C, indicating roles for both MyD88 and TRIF-TICAM-1 in NF-κ B activation through TLR4. This is supported by the observation that LPS fails to induce NF-κ B activation in fibroblasts from TRIF/TICAM-1–MyD88 double KO mice (34).
Recently, Hoebe et al. (35) identified a chemically induced mutation (Lps2) in mice that led to hyporesponsiveness to LPS. This mutation was identified as a single base-pair deletion in TRIF/TICAM-1 that is predicted to replace the C-terminal twenty-four amino acids with an unrelated eleven amino-acid sequence. Nonetheless, the mutated TRIF gene still has the potential to encode a protein that contains a TIR domain and an N-terminal region that could bind to signaling proteins, such as IRF-3, TRAF-6, and TBK-1 (36, 37). The Lps2 mutant mice also exhibited diminished production of IFN-α /β that was correlated with an inability to control virus replication. Experiments in mice that harbor both the MyD88 KO and the Lps2 mutation, revealed that both adapters normally contribute to LPS-induced TNF-α secretion. Additionally, the authors observed that when wild type, MyD88 KO, or TRIF/TICAM-1 mutant macrophages were treated with LPS, virtually all wild-type cells made TNF-α, while the response of MyD88 KO macrophages was minimal. Surprisingly, however, more than half of the TRIF/TICAM-1 mutant cells also made TNF-α. When treated with polyI:C, approximately half of the wild-type cells produced TNF-α, and MyD88 KO macrophages exhibited no loss of responsiveness. In contrast, none of the TRIF/TICAM-1 mutant cells responded to polyI:C. There is, however, an inconsistency between these findings (Figure 6 of 33) and the data shown in their first figure (33) in which the TRIF/TICAM-1 mutant macrophages failed to secrete detectable TNF-α in response to either LPS or polyI:C. This suggests that TRIF/TICAM-1 and MyD88 may differentially regulate events that lead to the release of TNF-α into culture supernatants.
TIRP/TRAM/TICAM-2
The most recently identified TLR adapter, called TIRP (38), TRAM (26), or TICAM-2 (39), is 235 amino acids in length, but shares sequence similarity most closely with TRIF/TICAM-1. Notably, both human and mouse TIRP/TRAM/TICAM-2 contain a cysteine (C117 in humans) at the position where other adapters and TLRs have a conserved proline, although an adjacent proline (P116) is present. Unfortunately, several independent groups conducted similar studies but obtained radically different results and conclusions. Bin et al. (38) found that TIRP/TRAM/TICAM-2 interacted with IL-1R, IL-1RAcP (IL-1R accessory protein), TRIF/TICAM-1, and TIRAP/Mal, but not with TLR2 or TLR4. TIRP/TRAM/TICAM-2 also associated with kinase-inactive IRAKs and with TRAF6. Bin et al. found that overexpression of TIRP/TRAM/TICAM-2 also activated an NF-κ B reporter, but not an IFN-β reporter construct, a finding that is at odds with more recent studies (26, 39).
Fitzgerald et al. (26) analyzed the possible role of TIRP/TRAM/TICAM-2 in signaling by various TLRs. Like TRIF/TICAM-1, TIRP/TRAM/TICAM-2 overexpression augmented IRF-3 and IRF-7 reporter activities and, like TRIF/TICAM-1, induced IRF-3 nuclear translocation, and resulted in the activation of IFN-β, RANTES, IP-10, and IFN-α 1/α 2 promoters, implying that NF-κ B was activated because these genes are NF-κ B–dependent. Immunoprecipitation studies revealed that TIRP/TRAM/TICAM-2 interacts with TRIF/TICAM-1, as well as IRF-3, IRF-7, and the IRF-3,7 kinases, IKKε, and TBK1. TIRP/TRAM/TICAM-2 C117H, but not the TIR alone or the P116H mutant, inhibited LPS- but not polyI:C-induced RANTES or IRF-3,7 promoter-driven reporter activation. In contrast, the TRIF/TICAM-1 TIR blocked both polyI:C and LPS-induced RANTES, indicating that TRIF/TICAM-1 regulates both TLR3- and TLR4-stimulated IRF-3, whereas TIRP/TRAM/TICAM-2 is TLR4-specific. TIRP/TRAM/TICAM-2 C117H also blocks LPS-induced NF-κ B signaling, but fails to block TLR2, 3, 7, 8, IL-1β, or TNF-α –induced NF-κ B. These findings were confirmed using an siRNA silencing approach and show that LPS (TLR4) signaling leading to RANTES reporter activity is inhibited by siRNA for both TIRP/TRAM/TICAM-2 or TRIF/TICAM-1, while only the latter inhibited TLR3 signaling (26), indicating that TIRP/TRAM/TICAM-2 is necessary for both arms of the TLR4 signaling pathway.
The TRIF/TICAM-1 DN blocked RANTES promoter activity induced by TIRP/TRAM/TICAM-2, but the converse was not true. This suggests that that TIRP/TRAM/TICAM-2 requires TRIF/TICAM-1. Immunoprecipitation experiments revealed that TIRP/TRAM/TICAM-2 interacts with TRIF/TICAM-1 and TIRAP/Mal, and that TRIF/TICAM-1 also interacts with TIRAP/Mal (26). TRIF/TICAM-1 and TIRP/TRAM/TICAM-2 also interact with the TIRAP/Mal P125H mutant. This finding may explain why the TIRAP/Mal P125H DN blocked both IFN-β and NF-κ B induced by LPS, whereas the TIRAP/Mal KO mouse still retained the ability to induce activation of both transacting factors (i.e., by sequestration of TRIF/TICAM-1 or TIRP/TRAM/TICAM-2). However, it is still not clear how the TIRAP/Mal inhibitory peptide blocked signaling unless it interferes with the formation of a signaling platform comprised of TRIF/TICAM-1 and TIRP/TRAM/TICAM-2. Fitzgerald et al. also confirmed that TLR4 interacts with TIRAP/Mal, TIRP/TRAM/TICAM-2, but not TIRP/TRAM/TICAM-2 C117H (26). This would suggest that the cysteine residue located within the TIRP/TRAM/TICAM-2 BB loop is necessary for its interaction with TLR4. Neither TIRAP/Mal nor TIRP/TRAM/TICAM-2 interacted with TLR3. Although Fitzgerald et al. also found that TIRP/TRAM/TICAM-2 was able to interact with the IL-1RAcP, they were unable to inhibit IL-1-induced NF-κ B using the TIRP/TRAM/TICAM-2 C/H DN or by siRNA silencing studies (K. Fitzgerald, unpublished).
In another very recent paper, Oshiumi et al. (39) largely confirmed the work of Fitzgerald et al. (26), but extended it significantly by providing additional critical data that assessed protein–protein interactions by yeast two-hybrid screening. They found that the direct interaction of TLR4 and TIRP/TRAM/TICAM-2 requires the proline residue in the TLR4 BB loop, but not the TIRP/TRAM/TICAM-2 cysteine that is in the analogous position. TRIF/TICAM-1 binds directly to TLR3, but not to TLR4, despite the fact that this latter interaction was detected in immunoprecipitation experiments. The formation of TIRP/TRAM/TICAM-2 homodimers depends upon the presence of the BB loop cysteine, as does the interaction of TIRP/TRAM/TICAM-2 with TRIF/TICAM-1. Finally, neither TRIF/TICAM-1 nor TIRP/TRAM/TICAM-2 bind to MyD88 or TIRAP/Mal in yeast two-hybrid experiments, in contrast to results obtained by immunoprecipitation. A TIRP/TRAM/TICAM-2 KO mouse has recently been generated (40). Consistent with the findings of Fitzgerald et al. and Oshiumi et al. (26, 39), this mutant mouse is profoundly LPS-unresponsive, yet retains polyI:C responsiveness.
What to Make of It All?
At this point, the reader is undoubtedly wondering “How does this all come together?” We believe that the model shown in Figure 2⇓ accounts for most of the published data with regard to TLR4 signaling, and we will only comment on signaling by other TLRs where useful comparisons can be made. Nonetheless, we recognize that new results will likely make it necessary to modify our model soon; however, the model should provide the reader with a basis for testing hypotheses based on existing data.
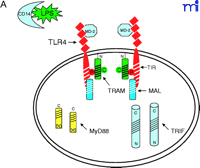
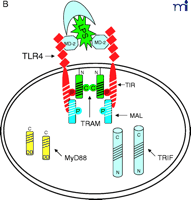
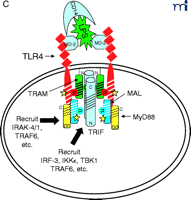
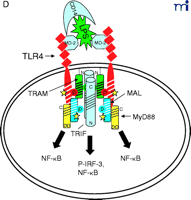
Stepwise model for TLR4 signaling by LPS. See text for descriptions of panels A–D.
The diagram in Figure 2A⇑ shows a cell that expresses TLR4 and various adapter molecules discussed above prior to stimulation with the TLR4 agonist LPS; in this model, LPS is shuttled to the cell by soluble CD14. MD-2, a necessary extracellular protein that enables TLR4 to respond to LPS (41), is associated with TLR4. Based on data obtained from direct protein–protein interaction studies, TLR4 interacts directly with TIRAP/Mal (“Mal” in Figure 2⇑) and with TIRP/TRAM/TICAM-2 (“TRAM” in Figure 2⇑) in an agonist-independent manner. The latter interaction requires the proline in the TLR4 BB loop (39). Mutation of a recently identified myristoylation site in the N terminus of TIRP/TRAM/TICAM-2 results in its dissociation from the membrane (K. Fitzgerald, unpublished); therefore, TRAM is depicted as tethered to the plasma membrane.
In response to LPS stimulation of the cell (Figure 2B⇑), the two TLR4 molecules are brought into juxtaposition, permitting homodimerization of TIRP/TRAM/TICAM-2 through their BB loop cysteine residues. This is consistent with the observations that this cysteine is essential for homodimerization and for binding TRIF/TICAM-1 (26, 39). We propose that the complex formed between the TLR4 molecules and TIRP/TRAM/TICAM-2 homodimer creates the main platform to which other adapters––MyD88 and TRIF/TICAM-1 (“TRIF” in Figure 2⇑)––are recruited (Figure 2C⇑). The requirement of TIRP/TRAM/TICAM-2 in this platform is suggested by data showing that TLR4-mediated signaling is dramatically reduced in TIRP/TRAM/TICAM-2 KO mice (40). MyD88 interacts directly with TIRAP/Mal (20), although a direct interaction of MyD88 with TLR4 has not been demonstrated. This makes our model radically different from previous models of TLR4 signaling. We suggest that the TLR4 BB loop proline is essential for TLR4 signaling, as evidenced by the LPS-unresponsiveness of the C3H/HeJ mouse TLR4 P/H mutant strain (42), because TLR4 P712 interacts with TIRP/TRAM/TICAM-2, and not with MyD88; however, this hypothesis will have to be tested formally. TRIF/TICAM-1 directly contacts TIRP/TRAM/TICAM-2 (consistent with the direct binding data; 39), and associates with TLR4 only indirectly. We have been able to detect tyrosine phosphorylation of TLR4 after stimulation with LPS (M. Fenton, unpublished results; and A. Medvedev and S. Vogel, unpublished results), and TIRAP/Mal also becomes phosphorylated upon LPS stimulation (L. O’Neill; unpublished results), although the precise sites of phosphorylation and the functional significance of these adapter modifications in LPS signaling are unclear. We believe that phosphorylation (indicated by stars in Figure 2C⇑) may be critical for the interactions amongst the molecules within the platform and enables subsequent recruitment of various kinases required for NF-κ B signaling (e.g., IRAK-1, IRAK-4, etc.) or for IRF-3 phosphorylation (e.g., IKKε, TBK1, etc.), leading to IFN-β gene expression. Again, the model accounts for the physical association of such proteins with the various adapters. It also accounts for the fact that MyD88 or TIRAP/Mal KO mice exhibit residual NF-κ B activation and IRF-3 phosphorylation, whereas TRIF/TICAM-1 KO mice exhibit somewhat depressed NF-κ B activation (probably mediated through the MyD88-dependent pathway) but lose IRF-3 activation and IFN-β inducibility. The model also predicts that the “Lps2 ” mutation, which results in a frameshift that affects expression of the terminal twenty-four amino acids of TRIF/TICAM-1 (35), must contribute in an important way to the interaction of this molecule with TIRP/TRAM/TICAM-2 or other proteins necessary for signaling. Not shown, but equally important, are the earlier findings of Perera et al. (43, 44) that both CD14 and CD11b/CD18 contribute in a significant way to the induction of distinct subsets of LPS-inducible genes. How these molecules interact with the TLR4 signaling complex to regulate expression of distinct gene subsets of genes remains to be elucidated.
The current data suggest that the interaction of TLR4 with TIRP/TRAM/TICAM-2 is unique in its utilization of all four known TLR adapter molecules, and that this leads to a more complex signaling pattern than is observed with other TLRs. However, TLR4 is probably the most closely studied of the TLRs and it remains possible that similar levels of complexity for the other TLRs are waiting to be discovered. The ability of TLR3 to interact directly with TRIF/TICAM-1 (39) and its apparent lack of dependence on MyD88 (Figure 3⇓), for example, would be predicted to facilitate a more robust IFN-β response––something that would be desirable because TLR3 is stimulated by double-stranded RNA during the course of viral infections. The failure of TLR2 to recruit TRIF/TICAM-1 or TIRP/TRAM/TICAM-2 (K. Fitzgerald, unpublished) is not surprising because most of the molecules that stimulate TLR2 are derived from extracellular, Gram-positive bacteria that are eliminated by IFN-independent mechanisms. Therefore, the possibility that TLR2 utilizes TIRAP/Mal and MyD88 only, as depicted in Figure 4⇓, is consistent with the mitigated capacity of TLR2 agonists to induce gene expression. At this time, the utilization of specific adapter combinations by other TLRs is less well-defined.
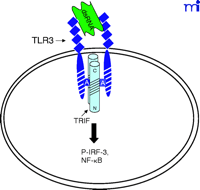
Model for TLR3 signaling by double stranded RNA (dsRNA). See text for detail.
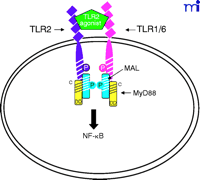
Model for TLR2 signaling by TLR2 agonists. See text for detail.
Given the differences in signaling between mammalian TLRs and their Drosophila orthologs, the MyD88-independent pathway mediated by TRIF/TICAM-1 and TIRP/TRAM/TICAM-2 appears to represent a more recent event in molecular evolution. Evolutionary pressure to create a MyD88-independent host response may have arisen as a consequence of the need to expand the capacity of mammals to defend against specific pathogens (e.g., Gram-negative bacteria). The TLR4-dependent activation of IRF-3 (45, 46), that leads to production of IFN-β, activation of STAT1, and induction of STAT1-dependent genes (24), represents a major advance in the potency and sophistication of the innate immune response compared with responses induced by TLR2 in mammals and by Toll in Drosophila. Finally, the complexity of TLR4 signaling is underscored by the fact that gene expression in response to LPS signaling is consistently more robust and diverse than is seen in response to other TLR agonists (3–5).
In summary, the ability of individual TLRs to discriminate between invading pathogens is an important determinant of the unique gene expression profiles activated in the host by different microorganisms. The outcome of pathogen recognition is critically dependent on the TLR-restricted utilization of TIR domain-containing adapter molecules, alone or in combination, to drive stimulus-specific responses.
Acknowledgments
The authors wish to extend their grateful thanks to Dr. Ian Sabroe for his insightful discussions during the preparation of this manuscript and to Dr. Daniel Caffrey for his valuable assistance in the development of Figure 1B⇑. This work was supported in part by NIH grants AI-18797 (SNV) and AI-47233 (MF). KAF is funded by the Wellcome Trust (London, UK).
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Matthew J. Fenton, PhD, is a Professor of Medicine in the Pulmonary and Critical Care Division, Dept. of Medicine, and holds a secondary appointment in the Department of Microbiology and Immunology. Dr. Fenton recently moved to UMB from Boston University Medical School.

Katherine A. Fitzgerald, PhD, is a Research Assistant Professor, Department of Infectious Diseases, University of Massachusetts Medical School, Worcester, MA and is the recipient of a Wellcome Trust International Research Fellowship.

Stefanie N. Vogel, PhD, is a Professor of Microbiology and Immunology, with a secondary appointment in the Pulmonary and Critical Care Division, Dept. of Medicine, University of Maryland, Baltimore (UMB). Dr. Vogel recently moved to UMB from Uniformed Services University of the Health Sciences. E-mail: svogel{at}som.umaryland.edu; fax (410) 706-8607.

