Galanin in Alzheimer Disease
- Scott E. Counts1,
- Sylvia E. Perez1,
- Stephen D. Ginsberg2,
- Sonsoles de Lacalle3 and
- Elliott J. Mufson1
- 1Department of Neurological Sciences, Rush-Presbyterian-St. Luke’s Medical Center, 2242 West Harrison Street, Chicago, IL 60612,
- 2Center for Dementia Research, Nathan Kline Institute, Departments of Psychiatry and Physiology & Neuroscience, New York University School of Medicine, 140 Old Orangeburg Road, Orangeburg, NY 10962
- 3Department of Biological Sciences, California State University, Los Angeles, 5151 State University Drive, Los Angeles, CA 90032
Abstract
Galanin (GAL) and GAL receptors (GALR) are overexpressed in limbic brain regions associated with cognition in Alzheimer disease (AD). The functional consequences of this overexpression are unclear. Because GAL inhibits cholinergic transmission and restricts long-term potentiation in the hippocampus, GAL overexpression may exacerbate clinical features of AD. In contrast, GAL expression increases in response to neuronal injury, and galaninergic hyperinnervation prevents the decreased production of protein phosphatase 1 subtype mRNAs in cholinergic basal forebrain neurons in AD. Thus, GAL may also be neuroprotective for AD. Further elucidation of GAL activity in selectively vulnerable brain regions will help gauge the therapeutic potential of GALR ligands for the treatment of AD.

Introduction
Galanin (GAL) is distributed throughout the mammalian central nervous system (CNS), where it modulates several ascending neurotransmitter systems, including the cholinergic, noradrenergic, serotoninergic, and neuroendocrine pathways (1–9) . Three G protein–coupled GAL receptors (GALR1, GALR2, and GALR3) signal in a tissue- and cell-specific manner to elicit a wide array of biological and behavioral responses (10–13) .
An important behavioral consequence of GAL activity is the regulation of cognitive function by GALR-mediated actions in the basal forebrain, entorhinal cortex, hippocampus, and amygdala (1 , 2 , 14–26) . In particular, GAL regulates the activity of cholinergic basal forebrain (CBF) neurons that provide the major cholinergic innervation of the cortex and hippocampus (27) and play a key role in memory and attention (15 , 26 , 28–30) . CBF neurons undergo selective degeneration during later stages of Alzheimer disease (AD) that correlates with disease duration and degree of cognitive impairment (31) . We and others have made the striking observation that GAL-containing fibers within the CBF undergo hypertrophy and hyperinnervate surviving CBF neurons in end-stage AD (32–34) . GAL levels are increased throughout the cortex in AD, and GALR binding sites are amplified in the CBF, hippocampus, entorhinal cortex, and amygdala during the course of the disease (20 , 23 , 35–37) . However, the role that GAL overexpression plays in AD is still unclear. GAL inhibits acetylcholine (ACh) release in rodent hippocampal preparations and disrupts cognitive performance in animals (1 , 2 , 15 , 19 , 26 , 38–40) , suggesting that GAL overexpression in AD basal forebrain may hamper ACh-mediated functions of remaining CBF neurons. Hence, excess GAL may exacerbate the cholinergic deficit seen in end-stage AD. On the other hand, GAL promotes neuritogenesis following sensory neuronal injury (41–43) , raising the possibility that increased GAL promotes neuronal survival to combat AD. This review focuses on galaninergic plasticity in brain regions that are associated with cognition in AD and on the therapeutic potential of GALR ligands for the treatment of this debilitating disease.
Galanin
GAL is a twenty-nine–residue (thirty in humans) peptide cleavage product of preprogalanin (44–46) . The preprogalanin gene has been cloned from several species (47–49), and its promoter contains binding sequences for regulatory factors whose activities are regulated by nerve growth factor (NGF), estrogen, protein kinase C, and adenosine 3’,5’ monophosphate (cAMP) (50–52) . The N-terminal fifteen amino acids of GAL are highly conserved in all species examined—from insects to humans—and are sufficient for high-affinity receptor binding (53–57) . Metabolic studies indicate that the GAL C terminus protects the N-terminal portion from proteolytic attack, resulting in increased bioavailability (58 , 59) .
GAL-immunoreactive (-ir) profiles have been described throughout the mammalian CNS in numerous species (34 , 60–66) . For example, in the mouse brain, GAL-ir neurons and fibers are found in several brain structures important for cognition, including: the cerebral cortex; the lateral and medial septum; vertical and horizontal limbs of the diagonal band of Broca and nucleus basalis of Meynert; the central and medial amygdaloid nuclei; and in the dentate gyrus and CA3 regions of the hippocampus (61 , 65) (Figure 1⇓).
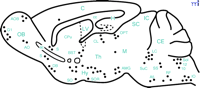
Schematic representation of a sagittal section of the mouse brain showing the distribution of GAL immunoreactivity in neurons (filled circles). A5, A5 noradrenergic cells; ac, anterior commissure; AMG, amygdala; AO, anterior olfactory nucleus; AOB, accessory olfactory bulb; BST, bed nucleus of the stria terminalis; C, cerebral cortex; cc, corpus callosum; CE, cerebellum; CL, central lateral thalamic nucleus; CPu, caudate-putamen complex; DB, diagonal band of Broca; G1, glomerular layer of the olfactory bulb; H, hippocampus; Hy, hypothalamus; IC, inferior colliculus; IO, inferior olive; LC, locus coeruleus; LV, lateral ventricle; M, mesencephalic tegment; OB, olfactory bulb; OPT, olivary pretectal nucleus; RF, reticular formation; S, septum; SC, superior colliculus; SO, supraoptic nucleus; Sol, solitary tract nucleus; SOR, retrochiasmatic supraoptic nucleus; SS, superior salivatory nucleus; SuC, subcoreuleus nucleus; Th, thalamus; 10, dorsal motor nucleus of vagus nerve; 12, hypoglossal nucleus.
Several lines of evidence suggest that GAL inhibits neurotransmission in the CNS. GAL promotes nociception by attenuating the C-fiber transmission that underlies pain sensation (67 , 68) . Electrophysiologic studies show that GAL reduces the firing rate of noradrenergic locus coeruleus neurons by increasing their K+ conductance (6 , 9 , 69) . GAL hyperpolarizes cell membranes of serotoninergic dorsal raphe neurons (8) , and GAL infusion into the dorsal raphe nucleus decreases the expression of tryptophan hydroxylase and reduces serotonin release in the ventral hippocampus (3 , 70) . GAL reduces dopamine release from the ventral tegmental area and median eminence (71–73) , due possibly to a GAL-mediated opening of G protein–gated inwardly rectifying K+ channels (GIRKS) and a concomitant reduction in tyrosine hydroxylase expression (74) . Finally, as detailed below, GAL inhibits the evoked release of ACh and glutamate in rodent hippocampus (2 , 18 , 75–78) .
Galanin Receptors
The activity of GAL in the CNS is mediated by at least three GALR subtypes termed GALR1, GALR2, and GALR3 [for reviews see (10–13) ]. GALRs are members of the integral membrane, rhodopsin-like G protein–coupled receptor superfamily. GALR subtypes show substantial differences in their primary sequence, distribution, and functional coupling that may contribute to a diverse repertoire of signaling activities in various CNS pathways. Table 1⇓ summarizes GALR subtype-specific expression patterns in brain regions associated with cognition, and Table 2⇓ describes the in vitro activities of the cloned receptors. In general, GALR1 activation reduces cAMP concentrations, opens GIRK channels, or stimulates mitogen-activated protein kinase (MAPK) activity in a pertussis toxin (PTX)-sensitive manner, consistent with GALR1 coupling to Gi/o proteins (79–82) . GALR2 activation increases inositol phosphate hydrolysis, mediates the release of Ca2+ into the cytoplasm from intracellular stores, and opens Ca2+ -dependent chloride channels in a PTX-resistant manner, indicating that GALR2 couples to Gq/11 proteins (80 , 82–86) . GALR3, like GALR1, elicits an inhibitory response by opening GIRK channels in a PTX-sensitive manner (81 , 85) . Future research aimed at understanding the distribution and activities mediated by endogenous GALR subtypes will be critical for gauging the therapeutic efficacy of various GALR ligands for the amelioration of AD symptoms. The three GALRs are also found extensively in peripheral tissues, including the digestive tract (79 , 85 , 87) , which may pose contraindications for drugs designed to modulate GALR activity in the CNS. Moreover, membrane binding studies demonstrating high affinity binding of GAL(1–15) but not full-length GAL in rat hippocampus (88–90) , and high affinity binding of GAL(3–29) but not GAL(1–15) in rat anterior pituitary (91), suggest the presence of unidentified GALR(s).
GALR mRNA Expression in Brain Regions Associated with Cognition
Putative Signaling Mechanisms of GALR Subtypes in Vitro
Galanin in Alzheimer Disease
GAL-ir fibers enlarge and hyperinnervate surviving CBF neurons within the septal diagonal band complex and nucleus basalis of patients with end-stage AD (32–34) (Figure 2A and B⇓). Using confocal laser microscopy, we demonstrated terminal appositions between GAL-containing fibers and CBF neurons in the normal human brain and that these contacts hypertrophied in AD (32) . Recently, we determined via a two-site ELISA assay that GAL concentrations in the nucleus basalis of pathologically confirmed AD subjects were increased ∼3-fold in the AD nucleus basalis compared to GAL concentrations in age-matched controls (Mufson, unpublished observations), supporting an earlier study showing a ∼2-fold GAL increase in AD basal forebrain (92) . This data corroborates light microscopic studies of GAL overexpression in the AD basal forebrain and lend support to the concept that GAL may regulate CBF tone in end-stage AD. To understand the role of GAL overexpression during the development of AD, our group has undertaken a semi-quantitative study of GAL-ir profiles in the anterior nucleus basalis of people clinically diagnosed with mild cognitive impairment or mild AD (93) . The anterior nucleus basalis shows the greatest degree of GAL hyperinnervation in the CBF in end-stage AD (32–34) . Preliminary data from these investigations indicate that GAL fibers do not hyperinnervate this region of the CBF during the prodromal or early stages of AD. This finding suggests that GAL overexpression in the CBF occurs mainly during later stages of the disease process.
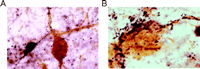
GAL plasticity in the basal forebrain nucleus basalis in AD. A. Photomicrograph shows a magnocellular cholinergic nucleus basalis neuron immunostained with the CBF neuronal marker p75NTR (brown reaction product) and innervated by GAL-ir fibers (dark blue reaction product) in aged control brain. The GAL-ir parvicellular neuron contacting the CBF neuron is visible. B. The photomicrograph shows striking hyperinnervation of GAL fibers impinging upon a nucleus basalis CBF neuron in AD.
Additional evidence for GAL plasticity comes from GAL radioimmunoassays (RIA) and GALR binding studies in aged-but-intact or AD brains. Gabriel and coworkers (35) demonstrated a ∼20–60% increase in GAL in frontal, temporal, and parietal cortical association areas in AD, but not in patients with schizophrenia. The distribution of GALR binding sites was studied in the normal post-mortem human brain using [125 I]-hGAL in combination with in vitro receptor autoradiography (94) . This study demonstrated high binding density in the basal forebrain. GALR binding sites were also detected in all cortical areas and in layer II of the entorhinal cortex, the uncus, and the hippocampal-amygdala transition area (94) . Our group studied the expression pattern of GALRs in the nucleus basalis in pathologically defined early (mild) and late (severe) AD cases using quantitative in vitro autoradiographic imaging of [125 I]-hGAL binding sites (20) (Figure 3A–C⇓). The highest labeling density in the aged control nucleus basalis complex was found within the intermedioventral division, the lowest within the posterior division, and an intermediate density was seen within the anterior regions. GALR labeling in age-matched early AD cases was similar to that found in the controls. Significantly, the density of GALRs in the anterior region of the CBF increased in late AD compared to early AD or aged controls (Figure 3A–C⇓). Interestingly, the hyperinnervation of GAL-containing fibers upon surviving CBF neurons in end-stage AD occurs primarily in the anterior CBF (32–34) , which is relatively spared in AD (95 , 96) . On the other hand, the posterior nucleus basalis, which undergoes severe degeneration in AD (95 , 96) , displayed the least [125 I]-hGAL binding in control or AD subjects (20) . These findings suggest a putative positive effect of GAL overexpression on CBF neuron survival in AD.

Plasticity of GAL binding sites in early and late stage AD. Autoradiograms showing the regional distribution of [125I]-hGAL binding to GALRs in the aged control (A, D) brain as compared to early (B, E) and late stage (C, F) Alzheimer disease. Pseudocolor density maps show changes in [125I]-hGAL labeling in the basal forebrain (A–C), entorhinal cortex (D–F), and amygdala (D–F) during the progression of the disease. There is an increase in labeling in the anterior subfield of the nucleus basalis in late AD (compare C with A and B). In contrast, the entorhinal cortex displays increased expression of GAL binding only in early AD (compare E with D and F). Likewise, GAL binding increases dramatically in the central nucleus and cortico-amygdaloid transition area of the amygdala in early AD and returns to levels similar to controls by late AD. AB, accessory basal nucleus of the amygdala; ac, anterior commissure; BL, basolateral nucleus (amygdala); BNST, bed nucleus of the stria terminalis; CAT, cortico-amygdaloid transition area; Ce, central nucleus (amygdala), Ch4a, anterior cholinergic cell groups of the nucleus basalis; Co, cortical nucleus (amygdala); ENT, entorhinal cortex; Gp, globus pallidus; ic, internal capsule; L, lateral nucleus (amygdala); Pt, putamen. Colorimetric scale (inset) correlates pseudocolor density with [125I]-hGAL binding density.
GALR binding studies demonstrate that galaninergic plasticity occurs in the entorhinal cortex, hippocampus, and amygdala in AD. [125 I]-hGAL binding sites have been found in cortical layers II and IV of the human entorhinal cortex (36 , 94) , which plays a crucial role in the transfer of feed-forward cortico-cortical information related to memory (97) . This is intriguing because the layer II neurons provide the major glutamatergic excitatory input to the hippocampus that travels within a fiber bundle known as the perforant pathway and degenerates very early in AD (97–99) , whereas layer IV receives sigificant hippocampal efferent projection (97) . Our in vitro autoradiography studies of [125 I]-hGAL binding in control and AD subjects revealed a ∼3-fold increase in GALR binding sites in entorhinal cortex layer II in early AD patients compared to those with late AD, when binding levels were increased only slightly over control (23) (Figure 3D–F⇑). Similarly, Rodriguez-Puertas and colleagues (37) demonstrated that GALR binding sites increased ∼10–30% in the entorhinal cortex and ∼50–100% in the hippocampus in late AD. [125 I]h-GAL binding sites are also found in the central nucleus and the cortico-amygdaloid transition area of the amygdala, which has reciprocal connections with the basal forebrain, hippocampus, and cortex (e.g., superior temporal and entorhinal cortices), plays a pivotal role in higher order cognitive processing, and displays extensive AD-related pathology early in the disease process (23 , 100–102) . [125 I]-hGAL binding is increased in these same areas of the amygdala in early/probable AD but not during late-stage AD (23) (Figure 3D–F⇑). Whether increased GALR expression in limbic and cortical areas related to cognitive function represents an attempt at rescuing these neuronal populations remains to be determined.
Potential Triggers Of GAL Plasticity In AD
The pathophysiological factors that induce GAL plasticity in the AD brain have been a matter of great speculation. In general, the spatio-temporal pattern of GAL overexpression coincides with the degeneration of select neuronal populations during the course of the disease. Limbic areas (e.g., entorhinal cortex and amygdala) that degenerate in the prodromal stage of AD (98 , 99 , 102) are sites that overexpress GAL early in disease (23) , whereas the plastic response occurs during the later stages of AD within the cholinergic system (20 , 32–34) . A parsimonious explanation for these phenomena is that GAL systems hypertrophy in response to neuronal injury. In this regard, observations that GAL plasticity does not occur in the nucleus basalis during the prodromal stage of AD (93) when CBF neurons are preserved (103) suggest that GAL hyperinnervation of the nucleus basalis in end-stage AD is an attempt to rescue degenerating CBF neurons and to maintain cholinergic function. Recently, we tested whether neurochemical lesions of CBF neurons of the horizontal limb–diagonal band (HDB) by the selective cholinergic cell toxin 192IgG–saporin would induce GAL hyperinnervation of this CBF subfield (16) . The immunotoxic lesion caused a ∼30% loss of cholinergic cells ipsilateral to the lesion. Increased GAL immunoreactivity with a thickening of GAL-ir fibers in both ipsilateral and contralateral HDB subfields was observed as early as one hour and as late as twenty-four weeks after the lesion (Figure 4⇓). The number of cholinergic neurons did not recover to control levels after six months, nor did the density of GAL immunoreactivity decrease to control levels in that same period. These findings of increased local GAL following neurochemical lesioning of the HDB by either the selective cholinergic cell toxin 192IgG–saporin (16) or ibotenic acid (104) support the notion that GAL plasticity is triggered by neuronal damage.
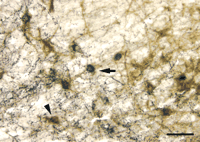
GAL innervation of basal forebrain HDB following immunolesion with the selective cholinergic cell toxin 192IgG–saporin. Photomicrograph of ipsilateral lesioned HDB shows p75NTR-ir CBF neurons (brown reaction product; arrowhead) and p75NTR-ir CBF neurons containing heavy deposits of GAL immunoreactivity (dark blue reaction product; arrow). The beaded and punctate dark blue deposits resembling GAL-ir axons and terminals covering some of the p75NTR-ir dendrites are visible. Scale bar = 50 mm.
AD neurodegenerative lesions may also play a role in GAL hypertrophy. Human neuropathological studies have shown that neuritic plaques contain GAL (105) , whereas neurofibrillary tangle-bearing neurons do not contain appreciable levels of GAL immunoreactivity (Mufson, unpublished observations). Studies using transgenic mice that overexpress human amyloid precursor protein bearing the AD-related V717F mutation suggest a role for plaque deposition in GAL overexpression (106) . Older mice exhibit age-related increases in amyloid plaque deposition in the hippocampus and entorhinal cortex and display a prominent up-regulation of GAL-ir fibers into these areas. Dystrophic GAL-ir neurites are found in many of the amyloid plaques, and occasional GAL-ir cell bodies can be observed in the hippocampus that are not evident in wild-type mice (106) . The hippocampus and entorhinal cortex contain extensive plaque deposition and are first affected in AD (98 , 107) ; therefore, amyloid deposition might trigger the overexpression of GAL in these areas in early stages of the disease.
A final factor underlying GAL plasticity in AD may be related to ApoE genotype, the major genetic risk factor for late-onset sporadic AD (108–110) . Comparison of GAL concentrations in the nucleus basalis between AD subjects carrying at least one apoE4 allele (apoE3,4 or apoE4,4) and AD subjects lacking an apoE4 allele revealed a trend (p = 0.12) for a twofold increase in GAL concentration in the nucleus basalis in the presence of apoE4 (Mufson, unpublished observations). ApoE4 gene dosage is inversely related to choline acetyltransferase (ChAT, the synthetic enzyme for ACh) activity and to ACh binding levels in the hippocampus and temporal cortex of AD cases (111 , 112) . Moreover, the presence of apoE4 is associated with reduced efficacy of cholinesterase inhibitor therapy in AD (112) . Thus, the influence of apoE genotype on cholinergic function may contribute to GAL overexpression in the basal forebrain in AD.
Neuronal Origin of GAL Hyperinnervation in AD
The source(s) of GAL hyperinnervation and GALR plasticity in AD remain unclear. For instance, it is unlikely that the few small GAL-ir neurons within the basal forebrain and preoptic area account for the rich galaninergic fiber plexus seen within this region (33 , 34 , 60 , 62) . One potential source of GAL fiber innervation to the basal forebrain may be the locus coeruleus (113 , 114) . The coeruleo-forebrain pathway is well characterized in the mammalian CNS (115) , and GAL-ir cells within the locus coeruleus also exhibit enhanced GAL immunoreactivity in AD (116) . However, the human locus coeruleus does not appear to contain numerous GAL-ir cells (62) . Another source of GAL fibers may emanate from the central nucleus of the amygdala and course through the basal forebrain en route to the substantia innominata, bed nucleus of the stria terminalis, and hypothalamus (34) . Although the cells of origin of this GAL-ir bundle are undetermined, the input may arise from the extended amygdaloid complex, which contains numerous GAL-ir cell bodies (34 , 117) and displays hypertrophy of GAL-ir fibers in AD (Mufson, unpublished observations).
GAL Plasticity As A Detrimental Factor In AD
The functional consequences of GAL plasticity in AD are of intense interest. The most compelling evidence that GAL overexpression exacerbates the presentation of AD comes from rodent studies showing that GAL inhibits ACh release in the hippocampus and disrupts cognitive performance (1 , 2 , 19 , 26 , 38–40) . GAL inhibits the evoked release of ACh in the ventral hippocampus of the rat in a concentration-dependent manner and blocks the slow cholinergic excitatory post-synaptic potential (EPSP) induced by the release of endogenous ACh onto CA1 pyramidal neurons (1) . Furthermore, administration of GAL into the medial septum–diagonal band complex or ventral hippocampus impairs cognitive performance on several spatial learning and working memory tasks in rats (19 , 26) . GAL interference of cholinergic transmission during these tasks is particularly evident in the presence of muscarinic ACh receptor antagonists or cholinergic immunotoxin lesions (38 , 40 , 77) .
A role for GAL in glutamate-mediated long-term potentiation (LTP) in the hippocampus may also contribute to GAL’s effects on memory. Electrophysiological studies show that GAL restricts LTP at both perforant path-dentate gyrus and Schaffer collateral-CA1 synapses (14 , 18 , 22 , 24) . GAL may impact glutamatergic transmission in the hippocampus by reducing evoked glutamate release (18 , 75 , 78 , 118) . However, a recent study demonstrated that while GAL inhibits LTP at CA1 synapses, it has no effect on ionotropic α -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) or N -methyl-D-aspartate (NMDA) glutamate receptor-mediated EPSPs, suggesting that GAL acts through a postsynaptic GALR to inhibit LTP-related signaling cascades (14) .
The development of transgenic mice that overexpress murine GAL under control of the dopamine β -hydroxylase promoter (GAL-tg) has facilitated the study of GAL overexpression in the brain (25 , 119) (Table 3⇓). GAL-tg mice display increased GAL fiber density in the basal forebrain and a ∼3-fold reduction in the number of ChAT-ir neurons in the HDB subfield (25) . In situ hybridization experiments demonstrated a down-regulation of ChAT mRNA per cell within the HDB without a difference in the number of ChAT mRNA-containing HDB cells in GAL-tg mice (119) , suggesting that GAL overexpression in the basal forebrain of GAL-tg mice selectively reduces the expression of the cholinergic neuron phenotype. Spatial navigation testing with the Morris water task in GAL-tg mice showed a complete lack of selective search on the probe trial at eight, sixteen, and twenty-four months of age (25) . GAL-tg mice displayed normal swim speed, swimming patterns across time bins, and visible and hidden platform acquisition. Thus, the GAL-tg mice could perform all of the procedures in the Morris water maze but could not generate a cognitive map of the spatial environment to solve the probe test, the most challenging component of the this task (25 , 119) . The Morris task requires an intact, functioning hippocampus (25) , thus, it seems likely that the mechanisms underlying the observed deficits in GAL-tg mice include inhibitory neuromodulation by GAL in the hippocampus. Along these lines, GAL expression is increased as much as ∼4-fold in the hippocampus of GAL-tg compared to wild-type (WT) mice (120) , and GAL overexpression reduces glutamate release and restricts LTP at perforant path-dentate gyrus synapses in GAL-tg hippocampal slices (18) . Similarly, transgenic mice that overproduce GAL expressed from a platelet-derived growth factor B promoter (GalOE) had a ∼4-fold increase in hippocampal GAL and reduced frequency facilitation of field EPSPs—a form of short term synaptic plasticity—at mossy fiber-CA3 synapses in GalOE hippocampal slices (121) (Table 3⇓).
Characteristics of GAL and GALR Mutant Micea
Altogether, these data suggest that the GAL overexpression that occurs in the AD basal forebrain, hippocampal formation, and cortex acts to coordinate the inhibition of multiple neurotransmitter systems involved in cognitive function.
GAL Plasticity As A Neuroprotective Factor In AD
An alternative hypothesis is that GAL is neuroprotective in AD. A role for GAL in cholinergic cell survival was demonstrated in a knockout mouse model carrying a targeted loss-of-function mutation in the GAL gene (GAL-KO) (13 , 22) (Table 3⇑). GAL-KO mice show significant decreases in the number of ChAT-ir neurons in the basal forebrain medial septum (MS) and vertical limb–diagonal band (VDB) subfields. Moreover, these areas and the nucleus basalis displayed a significant decrease in the number of neurons expressing TrkA, the high affinity receptor for the cholinergic survival protein NGF (22) . Hence, GAL may act as a trophic factor for CBF neurons during development. On the other hand, preliminary data in our lab has demonstrated that the number of MS and VDB subfield neurons expressing the p75NTR neurotrophin receptor is similar in WT and GAL-KO mice (122) . Because p75NTR is a well-established cholinergic marker (123) , these findings suggest that GAL is required for regulating the cholinergic phenotype of basal forebrain neurons during development. GAL-KO mice exhibit age-related decreases in evoked ACh release in the hippocampus, inhibition of LTP in the CA1 region of the hippocampus, and age-dependent behavioral decline in the Morris water maze (22) , indicating an excitatory role for GAL in these animals. Significantly, a recent electrophysio-logical study using rat primary basal forebrain diagonal band cultures showed that exogenous GAL reduced an array of inhibitory K+ currents in cholinergic neurons (17) . GAL also increased the excitability of these cells under current-clamp conditions (17) . Thus, GAL overexpression in AD may improve cognition by promoting the survival or cholinergic tone of CBF neurons and by preserving hippocampal LTP.
Additional findings support a role for galanin in neuronal repair. GAL expression increased ∼120-fold following peripheral nerve injury in dorsal root ganglia (DRG) preparations (43) . GAL promotes neuritogenesis in cultured DRG explants of WT mice and rescues a deficiency in neurite outgrowth observed in DRG explants of GAL-KO mice (41 , 42) . GAL mRNA and protein are also increased near lesions in several rat models of CNS degeneration, including decortication (i.e., cortical transection), lesioning of the entorhinal cortex and ventral hippocampus (124 , 125) , lesioning of the CBF (16 , 104) , and spinal cord axotomy (126) . Thus, increased GAL expression may occur in vulnerable brain regions in response to neuronal damage. Notably, GAL hyperinnervation and GAL binding sites in AD are greatest in the anterior subfields of the basal forebrain where the least amount of neural degeneration occurs, whereas GAL overexpression is least found in the posterior subfields of the basal forebrain where degeneration is greatest (20 , 33 , 95 , 96) .
We have used single cell RNA amplification technology (127–130) to address the genetic consequences of GAL hyperinnervation upon CBF neurons (131) . Using postmortem brain tissue from cognitively intact and end-stage AD subjects, single anterior nucleus basalis neurons immunopositive for the cholinergic p75NTR in the presence or absence of hyperinnervating GAL-ir fibers [p75NTR (GAL+ ) or p75NTR (GAL– ), respectively] were microaspirated and processed for linear RNA amplification. Radiolabeled RNA probes generated from single cells were hybridized to custom-designed cDNA arrays (127–130) (Figure 5A⇓). Expression of protein phosphatases PP1α and PP1γ mRNA in p75NTR (GAL– ) neurons from AD brains was inhibited compared to phosphatase expression in p75NTR (GAL– ) neurons from control brains (Figure 5B). However, GAL-hyperinnervated p75NTR (GAL+ ) neurons do not display decreased expression of PP1α and PP1γ mRNA in AD brain (Figure 5A, B). As the reduction of protein phosphatases has been associated with the evolution of neurofibrillary tangle formation (127 , 132 , 133) , this apparent maintenance of PP expression by GAL may be neuroprotective for CBF neurons. However, a thorough analysis of the genetic signature of p75NTR (GAL– ) and p75NTR (GAL+ ) neurons will provide a more complete picture of the effects of GAL on gene expression in this cell group.
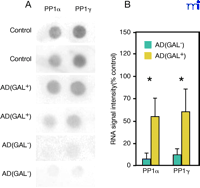
GAL innervation ameliorates down-regulation of protein phosphatase 1 (PP1) subtypes in AD nucleus basalis. Representative gene array (A) and histogram (B) shows a significant down-regulation of protein phosphatases PP1α (∼10% of control) and PP1γ (∼12% of control) mRNA in p75NTR(GAL–)-ir CBF neurons in AD nucleus basalis, whereas GAL-hyperinnervated p75NTR(GAL+)-ir CBF neurons do not have decreased expression of PP1α or PP1γ mRNA in AD. A total of twenty neurons were analyzed from normal control brains (n = 4 brains; 4–6 cells per brain); twenty-six neurons were analyzed from AD brains (n = 6 brains; 4–5 cells per brain) including 17 p75NTR(GAL–)-ir CBF neurons and 9 p75NTR(GAL+)-ir CBF neurons; *, p < 0.01 via ANOVA with Neuman–Keuls post hoc test for multiple comparisons.
The conflicting data regarding the nature of putative GAL overexpression in AD has yet to be reconciled, especially in light of the similar detrimental phenotypes observed in the GAL-KO and GAL-tg mice (Table 3⇑). Regarding the basal forebrain, results from the GAL-KO mouse suggest that GAL is important for the establishment of cholinergic basocortical and septohippocampal systems (22) . The hypertrophy of GAL-secreting neurons systems in AD may then represent a recapitulation of this developmental program in response to the cholinergic deafferentation of cortex and hippocampus. However, the potential compensatory effects of GAL plasticity may instead have deleterious consequences in the adult brain by inhibiting cholinergic transmission, as might be inferred from the phenotype of the GAL-tg mouse (25) . In any event, the roles of GAL activity in normal cognition and in AD remain undetermined. The development of transgenic animals overexpressing GALR subtypes or deficient for GALR subtypes may help to clarify the role(s) of GAL signaling in cognitive processes. For instance, initial reports on the development of GALR1 null mice (GALR1-KO) show that these mutants often exhibit severe spontaneous seizure activity in response to bright light or handling (134) (Table 3⇑). This observation suggests that GALR1 mediates GAL’s role in restricting glutamatergic transmission in the hippocampus and supports other data indicating that GAL may act as an anticonvulsant (18 , 135–137) .
GAL Receptors As Therapeutic Targets For AD
GALR1, GALR2, and GALR3 mRNAs are found in cell bodies of the rat basal forebrain (138–140) (Table 1⇑). However, very few cholinergic cells in the rat basal forebrain colocalize with GALR1 mRNA (117 , 141) , suggesting that GALR2, GALR3, or both, might mediate postsynaptic GAL effects on CBF neurons. If GALR2 is a postsynaptic receptor on CBF neurons, then augmented GAL input might be coupled to phospholipase C activation, the principal GALR2-mediated pathway identified in cell culture studies (80 , 82 , 84–86) (Table 2⇑). Activation of this pathway may improve cholinergic tone or survival of CBF neurons. Along these lines, the GALR2-specific agonist AR-M1896 (142) (Table 5⇓) promotes neurite outgrowth in GAL-KO DRG explants in a PKC-dependent manner, whereas a GALR1-specific antagonist RWJ-57408 (143) (Table 5⇓) was ineffective at blocking GAL-mediated neuritogenesis (42) . Alternatively, postsynaptic GALR2 or GALR3 may be coupled to adenylyl cyclase inhibition or GIRK activation (82 , 85 , 86) (Table 2⇑), potentially inhibiting cholinergic function. Finally, colocalization studies in rodents do not preclude the possibility that GALR1 is present on human CBF neurons. Future efforts addressing the protein expression patterns (e.g., pre- or postsynaptic) in human AD-related brain regions and the signaling repertoires of native GALR subtypes in these structures will be critical to understanding GAL activity in cognition and the efficacy of targeting GALRs for the treatment of AD.
Currently approved drug treatments for AD consist of cholinesterase inhibitors, which act by increasing the bioavailability of synaptic ACh. These drugs produce small but consistent improvements of memory and global function and positively influence activities of daily living (144–147) . If GAL inhibits ACh release, then GALR subtype-specific antagonists may enhance cholinergic transmission by reducing the inhibitory influence of GAL on the firing rate of CBF neurons. Likewise, if GAL promotes the survival or cholinergic tone of CBF neurons, then a GALR agonist might prove efficacious. Until recently, the only tools available for pharmacological differentiation of GALR subtypes have been synthetic GAL analogs with one or more amino acid substitutions or chimeric GAL peptide ligands that show variable affinity for human and rat GALRs and, incongruently, behave as antagonists at native receptors but as partial or weak agonists at cloned receptors (10 , 12 , 19 , 118 , 148–152) (Table 4⇓). However, two new peptidergic compounds, AR-M1896 and AR-M961, are selective agonists for GALR2 and GALR1–GALR2, respectively (142) (Table 5⇓). These compounds have been used to identify GALR subtype-specific activities in rat preparations, including nociception (mediated by GALR2) and analgesia (GALR1) in the spinal cord (140) , hyperpolarization of locus coeruleus neurons (GALR1) (6) , and neuritogenesis in DRG explants (GALR2) (42) . A limiting factor in the pharmacological characterization of GALRs has been the relatively slow discovery of potent and selective non-peptidergic agonists and antagonists. Four non-peptidergic compounds have been discovered that are either GALR1-specific agonists or GALR1-specific antagonists with variable affinities at human receptors (136, 143, 153–155) (Table 5⇓). Nonetheless, the field of GAL research is still developing, and the ongoing search for selective GALR ligands in drug discovery programs might provide new research tools for understanding GAL pharmacology and physiology. AD appears to arise from multiple etiologies, therefore a rational treatment strategy might include high-affinity GALR ligands used in combination with anticholinesterases and perhaps other compounds, such as cholinergic and glutamatergic agonists, anti-inflammatory drugs, or modulators of apolipoprotein E metabolism or function. We suggest that the pharmacological use of such compounds may ameliorate cholinergic hypofunction in AD and perhaps benefit other aspects of this heterogeneous disorder.
Ki of Nonspecific GALR Ligands at Cloned Rat GALRsa and Antagonist Actions of Chimeric GAL Peptides on Rat Behavioral Assaysb
GALR Subtype Specific Ligands
Conclusions
The presentation of AD is likely precipitated by neuronal degeneration in selectively vulnerable brain regions involved in higher cognitive processes. The GAL system plasticity in these regions during AD raises the intriguing possibility that pharmacological manipulation of GAL activity might be neuroprotective and slow the cognitive decline in AD. However, the functional consequences of GAL plasticity in AD must be clarified to guide the development of high-affinity GALR subtype-specific agonists or antagonists. Future elucidation of GALR distribution through the development of subtype-specific antibodies and of endogenous GALR activity through the development of subtype-specific ligands and transgenic GALR mice will be critical for gauging the therapeutic efficacy of GAL mimetics for the amelioration of AD symptoms.
Acknowledgments
The authors would like to thank our colleagues we have worked with over the years in exploring the nature of GAL plasticity in AD: M. Basile, W. Benzing, R.P. Bowser, J.N. Crawley, D.C. Deecher, I. Hartonian, J.H. Kordower, D.C. Mash, R.A. Steiner, and D. Wynick. Support for our research is provided by the following grants NIA AG10688, AG 09466, AG14449, TGAG000259, AG19597, NS43939, CA94520, and the Alzheimer’s Association.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Elliott J. Mufson, PhD , is the Alla V. and Solomon Jesmer Chair in Aging and Professor of Neurological Sciences at Rush-Presbyterian-St. Luke’s Medical Center, Chicago, IL. Sonsales de Lacalle, MD, PhD , is Assistant Professor of Biology at California State University, Los Angeles, CA. Stephen D. Ginsberg, PhD , is Assistant Professor in the Departments of Psychiatry, and Physiology and Neuroscience at New York University School of Medicine and the Center for Dementia Research, Nathan Kline Institute, Orangeburg, NY. Sylvia E. Perez, PhD , is Research Assistant Professor of Neurological Sciences at Rush-Presbyterian-St. Luke’s Medical Center, Chicago, IL. Scott E. Counts, PhD , is Research Assistant Professor of Neurological Sciences at Rush-Presbyterian-St. Luke’s Medical Center, Chicago, IL. Address all correspondence to EJM. Email: emufson{at}rush.edu; fax 312-563-3571.

