Protein Origami
Therapeutic Rescue of Misfolded Gene Products
- Oregon National Primate Research Center and Department of Physiology and Pharmacology Oregon Health & Science University 505 NW 185th Avenue Beaverton, OR 97006
- Address correspondence to PMC. E-mail connm{at}ohsu.edu; fax 503-690-5569.
Abstract
Receptor mutations that elicit loss of function are sometimes equated with defects that ablate receptor-ligand binding or receptor-effector interactions. Similarly, mutationally defective enzymes and ion channels are often viewed as compromised in substrate or ion recognition, respectively. Recent observations, however, suggest that an alternate mechanism may be surprisingly common, namely, that mutations in structural genes may not interfere with the inherent functionality of the affected protein, but nevertheless cause disease by preventing the cell’s trafficking machinery from placing the affected protein at the appropriate subcellular compartment (e.g., at the cell membrane). Accordingly, therapies may be devised to ensure the placement of receptors (or other proteins) at locations where they can support cell function.

Introduction
The regulation of cell function depends not only on the absolute number of receptors, ion channels, enzymes, and other proteins that are extant in the cell at any given moment, but also on the placement of these proteins within the cell. The endoplasmic reticulum is the site of synthesis and assembly of perhaps 100,000 proteins (counting heteromeric associations), the production of which is regulated at the translational and transcriptional levels by many factors, including ligands such as hormones, drugs, and growth factors. Moreover, as proteins are synthesized, they fold (1–4); this is no easy trick, given the proximity and diversity of proteins that accumulate in the cytosol to result in a nominal protein concentration of 100 milligrams per milliliter. Furthermore, the dihedral angles of the backbone appear to change synchronously in groups of four, thus avoiding dramatic steric interactions. As Cahill, Cahill, and Cahill observed of proteins, “They wriggle; they do not thrash” (5). The steric constraints that typify the protein backbone restrict the spectrum of protein shapes that must be recognized by the quality control apparatuses of the cell during protein routing and metabolism (6–7). Certain proteins may be routed by interactions with chaperones (8–11) or “sorting receptors” such as carboxypeptidase E (13). Mutations can result in defective protein conformations and in misfolding, misrouting, aggregation, and dominant negative forms (3–19). A list of diseases believed to involve such proteins is presented in Table 1⇓, although the data should be viewed as preliminary, consonant with the state of our understanding of this rapidly evolving area.
Disorders Believed to be Related to Misfolded Proteins
Processing of Receptors and Other Proteins
It is frequently assumed, although actual information is scarce, that as proteins are translated, they undergo posttranslational modification (e.g., addition of carbohydrate) and move with high efficiency to their target location. Recently, a view has been presented that the gene product that gives rise to the human delta opioid receptor is inefficiently processed, with only about half of the translation product reaching the membrane (20). The cellular expenditure of energy involved in this apparent inefficiency, however, may pay off by supplying the cell with a reserve of receptor molecules that can be shuttled to the surface when needed quickly (e.g., increased cellular sensitivity mediated by increased receptor number at the plasma membrane). The number of functional receptors—function being defined by position in the cell—can thus be tightly regulated.
Protein Misfolding and Routing in Disease States
Viewed as part of the processing by which proteins mature and become functional, the trafficking machinery contributes not only to the sorting and quality control of wild-type proteins, but may also act to dampen the effects of mutations—particularly mutations that interfere with the intrinsic capacity of proteins to mature and fold properly. Clearly, the protein sorting apparatus has important implications for disease (21–22), and also, to the degree that it can be equated with the accumulation of mutations, for aging (22). It has been argued that the practice of modern medicine (4, 23) promotes the accumulation of phenotypically silent mutations. Such mutations, in the absence of modern therapeutics, would otherwise be subjected to processes of natural selection.
Mutant proteins are often targeted for destruction. It is only recently that we have begun to appreciate the degree to which protein mutations—in particular, those that in no way interfere with the intrinsic functionality of the protein—may promote premature processing and destruction. Diseases that may arise as the consequence of mutant proteins that fail to fold properly may thus be treatable by approaches that would promote the proper folding and rescue of otherwise functional (albeit mutant) proteins.
Techniques to Correct Misfolded Receptors
Several approaches have been taken to rescue cells that produce defective proteins. Gene therapy that introduces wild-type sequences or acts to upregulate partially active mutant proteins has been an area of research for nearly a decade. Such approaches, however, are not of the type that we refer to when we discuss the “rescue” of mutant proteins. More specifically, the use of non-specific protein agents, such as polyols and sugars (25–27), has been described as a means of stabilizing protein conformations that may be particularly sensitive to denaturation. Most of us who handle dilute solutions of proteins add stabilizing agents, such as glycerol, to the solutions. Polyethylene glycols are used in purification of proteins for biotechnology applications and have received a great deal of attention in this regard. Although such approaches can rescue some mutant proteins, they are nonspecific in regard to rescuing a predesignated protein, and therefore, are of limited therapeutic value.
Genetic approaches to rescue mutant proteins by introducing compensatory modifications have also been described (33–35). These approaches either stabilize extant proteins rendered labile by genetic defects or usurp the trafficking machinery by adding targeting sequences. Although such genetic approaches can be highly specific to compensate for a given defective protein, they are probably redundant as therapeutic interventions, given that, were it possible to access the gene sequence, the primary error could be directly addressed. Secondary mutations that act to restore partial activity, however, are of considerable academic interest, because they demonstrate the possibility that primary mutations may not destroy ligand binding per se, but rather that they result in protein misfolding or misrouting.
Another corrective approach to proteins that are prone to misfolding would be to provide the cell with template molecules that would dispose nascent mutant proteins to adopting wild–type-like conformations and thereby promote their transport to their sites of function within the cell. We refer to this approach as “pharmacological rescue.” Desirable characteristics of a template molecule would include: 1) specificity for the molecule being rescued; 2) the ability to get to the right place (i.e., enter the cell, get to the endoplasmic reticulum, and associate effectively with the nascent protein); and 3) the ability to dissociate from the correctly folded protein (or, if not to dissociate, to allow the mature protein to function properly) after it arrives at the appropriate target locus. This third principle emphasizes that template molecules need not interact at the same site as the native ligand, a concept historically linked to the lessons of allosteric stabilization.
Proof of Principle: The GnRH Receptor
Gonadotropin releasing hormone (GnRH) plays a central role in neural regulation of reproductive function. This decapeptide is produced by specialized neurons found in the mediobasal hypothalamus, the axons of which project to the median eminence. From there, GnRH enters the portal circulation and binds to a specific receptor (i.e., GnRHR) on pituitary gonadotropes, stimulating synthesis and release of the gonadotropins LH and FSH. Sequence analysis of the GnRHR is consistent with the seven-transmembrane–domain motif, characteristic of the G protein–coupled receptor (GPCR) superfamily (31–33). The human GnRHR is coupled to the Gq/11 system; after GnRH binding, the activated GnRHR–Gq/11 protein complex activates the membrane-associated enzyme phospholipase Cβ, leading to the production of inositol 1,4,5-trisphosphate and the release of intracellular calcium (2).
Regulated Trafficking of the GnRH Receptor
Studies in comparative physiology of GnRHR suggest that nature has evolved specific means to restrict the availability of translated proteins for cellular function. In the case of the human GnRHR (328 amino acids; see Figure 1⇓), an additional amino acid, K191, is present (34, 35) relative to non-primate mammalian GnRHRs (327 amino acids). The additional lysine residue functions to lower the expression of the receptor at the plasma membrane in primates. Similarly, the cytoplasmic domains of GnRHRs from premammalian species, including fish and birds, have C-terminal extensions relative to mammalian forms of the receptor. The seventh transmembrane segment that typifies the mammalian GnRHR literally terminates at the cytoplasmic face of the plasma membrane, making it one of the shortest GPCRs. Recombinant constructs that fuse the fish/bird C-terminal extension to the mammalian GnRHR enhance expression of the receptor at the cell surface (34, 36). It thus appears that at least two translated signals within the GnRHR have evolved to regulate the surface expression of the receptor. Interestingly, the evolutionary differences in the primary structure of the GnRHR are accompanied by the evolution of two gonadotropins in mammals [i.e., luteinizing hormone (LH) and follicle stimulating hormone (FSH)], whereas fish have only a single gonadotropic hormone (i.e., GTH).
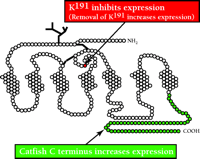
Two modifications of the GnRHR associated with altered expression. Addition of K191, an “extra” amino acid that appears in humans and other primates is associated with diminished receptor expression at the plasma, membrane. Pre-mammalian animals (fish, birds) have a C-terminal tail extension, absent in mammals, that enhances expression of the receptor. Accordingly, the tendency is to evolve a receptor that can be more closely regulated due to diminished expression.
Hypogonadotropic Hypogonadism: Disease as a Function of Protein Sequence
Hypogonadotropic hypogonadism (HH) is a human disease (although animal models are available for some forms) characterized by lack of development at puberty, incomplete or significantly delayed pubertal development, prepubertal testicular size into adolescence, and absence of secondary sexual development (e.g., pubic, facial, and auxiliary hair). HH has a number of causes, all of which interfere with GnRH signaling (37–45). Among the causes of HH are mutations of the GnRHR, which are almost always autosomal recessive (46–54). Predictably, patients with defects in the GnRHR respond poorly to exogenous GnRH.
At this time, fourteen different mutations to the human GnRHR have been described (46–52); thirteen result in single amino acid substitutions, and one results in deletion of fourteen residues from the C terminus. The fourteen mutations cause either complete or partial HH, and are generally distributed along the entire receptor sequence, including external, internal, and transmembrane domains (Figure 2⇓). When expressed in heterologous systems such as COS cells (which naturally lack a GnRHR), the mutant receptors, in comparison to the wild-type receptor, result in cells that are compromised in their ability to bind ligand, and cells that manifest surface expression of mutant receptors fail to transduce the hormone signal, consistent with the conclusion that the mutations interfere with ligand binding or effector coupling. An alternative conclusion is also possible, namely, that some mutations may result in protein misfolding so that the translation product is not routed to the cell membrane, where the mutant protein might prove to be fully functional. Recently, we investigated the merits of these alternative conclusions.
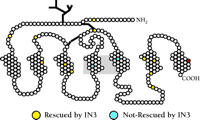
Pharmacological rescue of mutant GnRHRs. Loci of fourteen reported point mutations of the human GnRHR (N10K, T32I, E90K, Q106R, A129D, R139H, S168R, C200Y, S217R, R262Q, L266R, C279Y, Y284C, and L314X) isolated from patients with hypogonadotropic hypogonadism, of which eleven were rescued with IN3. The two internal substitutions that cannot be rescued by IN3 are less than 20 angstroms apart; the red circle represents the site of truncation of the C-terminal deletion mutant. The range of loci of mutation from which rescue can be effected is remarkably broad.
Engineered Modifications to Enhance Surface Expression of Mutant GnRH Receptors
Based on observations that come from comparative endocrinology, we explored two modifications of the GnRHR that are associated with altered expression at the plasma membrane. The first modification is based on the recognition (see above) that primate GnRHRs contain an extra lysine, relative to nonprimate species, at position 191 (Figure 1) and that this residue acts to limit expression levels (34, 36). The second modification that we pursued is based on recognizing that piscine GnRHR contains a C-terminal extension that, when fused to the mammalian receptor, increases receptor expression at the plasma membrane (35, 36). When both modifications are made to the human GnRHR, that is, deletion of the K191 and addition of the C-terminal tail, the enhancement of surface expression is greater than additive (35).
Accordingly, we selected one of the HH-associated mutant receptors (E90K) for detailed study. As expected, transfection of the gene that encodes this mutant receptor failed to confer hormone binding in COS cells, even though they clearly produced the mutant receptor. When either (or both) of the modifications described above were made to this mutant, however, the cells not only bound the hormone analog, but also effected receptor coupling as measured by inositol phosphate production (28). These observations showed that the E90K substitution did not interfere with receptor function; rather, that mutation had probably provoked misfolding of the gene product and prevented its placement at the cell surface. Consequently, we examined whether experimental interventions could result in more effective routing of mutant proteins so that they would become functional.
Pharmacological Rescue of Mutant Receptors
We reasoned that other mutations (see Figure 2⇑) of the human GnRHR resulting in HH might similarly result in protein folding defects that could be corrected, and we sought to identify a pharmacological agent that might effect rescue by serving as a folding template during translation. We regarded agonists as poor candidates as rescue agents, because they might provoke desensitization or downregulation. In addition, we sought a compound that was likely to enter cells. We selected a synthetic peptidomimetic antagonist of human GnRH (IC50 = 0.6 nM), produced by scientists at Merck & Co., Inc. (55–57), which we refer to as “IN3” (Figure 3⇓).
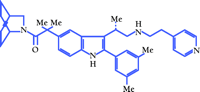
Structure of a GnRHR folding template. IN3 is the trivial designation of (2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-
oxoethyl]-2-(3,5-dimethylphenyl)-1H-indol-3-yl]-N-(2-pyridin-4-ylethyl)
propan-1-amine. Synthesized by Drs. Wallace T. Ashton and Mark Goulet (Merck & Co., Inc.).
For eleven of the fourteen HH-associated mutant receptors, addition of IN3 at the time of transfection conferred both ligand binding and effector coupling to transfected cells, showing that the effect of the mutational error could be corrected by the presence of the antagonist. The truncated mutant receptor, representing a comparatively severe mutation, proved to be incorrigible by IN3. In order to compare the behavior of the rescued E90K receptor with that of the wild-type GnRHR, we examined ligand specificity, an issue of pharmacological importance. Ligand specificity was indistinguishable from published values for the wild-type human receptor (22, 58–61), and both agonists and antagonists of GnRH were recognized by the rescued mutant receptor with responses characteristic of the wild-type receptor (60–62). The compelling finding that eleven of fourteen HH-associated mutant receptors could be rescued (as assessed by radioligand binding to cells and functional coupling to inositol phosphate production), despite widely disparate loci along the receptor molecule, offers the possibility that GnRHR mutants are frequently mispositioned although intrinsically functional (63).
Pharmacological Rescue: Protein Folding is Key
In order to address the degree of receptor distortion from which rescue could be effected, we also examined a palette of site-directed mutations in the human GnRHR at sites in which a cysteine normally appears, since this amino acid is associated with maintenance of tertiary receptor structure (59). When varied substitutions were made at the same locus (i.e., C278A, C278V, C278T, and C278M), there were differences in the ability to effect rescue. The mutants resulting from the more bulky substituents could apparently not attain the conformation necessary for rescue. Consistent with this view, it was not surprising to see that the expression of such variants on the plasma membrane also appeared variable in the absence of IN3. Substitution of the bulky Trp at position 279 with Ala (i.e., W279A) also produced an inactive mutant that was rescued by IN3.
Ala better accepted the folding template’s adjustment of configuration, resulting in the highest level of IP production. Mutation at C229 produced a defect from which rescue could not be effected, suggesting that this residue may have specific roles in receptor functionality. An alternative hypothesis, of course, is that this mutation results in a severe configurational insult that cannot be overcome by pharmacological rescue with IN3.
We also examined the ability to rescue deletion mutants. Deletion of the last three C-terminal amino acids results in loss of agonist-induced IP production; much of this activity could be recovered by IN3. Removing a larger (twelve amino acid) sequence from the C terminus produced a mutant that could not be rescued, whereas as many as four amino acids could be removed from the third intracellular loop (des237-241 GnRHR) and rescue still effected (59).
It is worth considering that, by virtue of the size of the peptidomimetic used in this study (i.e., IN3), a fairly small number of receptor residues appear to be involved in stabilizing conformation to the extent that IN3 can effect rescue. As a general matter, peptidomimetics of GnRH may be created by modeling the receptor-interacting sites that have been determined for GnRH. Other synthetic—and admittedly nonspecific—ligands of the GnRHR have been created based on commonality of the receptor to GPCRs as a superfamily. Still other peptidomimetics have been created by applying combinatorial chemistry to stable chemical nuclei, followed by high through-put screening. It would appear that, because the GnRH peptidomimetics are small molecules, they are able to create the stabilizing interactions. For some classes of peptidomimetics, there may be a relation between molecules that can occupy the active site and the critical amino acid interactions needed to stabilize the receptor. This observation may provide clues for the design of effective rescue molecules. On the other hand, molecules that function as rescue agents without mimicking ligand binding present the interesting possibility that agents may be identified that do not compete with ligand binding at all. The surface expression level of human wild-type receptor itself is enhanced by IN3 (59), presenting potential therapeutic approaches for disease states in which the wild-type receptor is processed excessively (e.g., targeted for proteasome degradation). Less than 100% of synthesized receptor is routed to the functionally active site on the plasma membrane.
Another Example of Pharmacological Rescue: Mutants of the V2 Vasopressin Receptor
Of the 150 or so mutations of the V2 vasopressin receptor gene known to cause nephrogenic diabetes insipidus, a large number of these mutant receptors are potentially functional but fail to fold properly and therefore are not routed to the cell surface (64). Bouvier and colleagues have used a nonpeptidic V2R antagonist to increase surface expression and rescue the function of eight of these mutants by promoting their proper folding.
The Other Side of the Coin: Shipwrecking Receptors
If it is possible to use a template such as IN3 to influence the correct shaping of a receptor, it is also possible that other compounds exist which cause the misfolding of a nascent receptor, driving it to an incorrect locus or resulting in degradation. We have chosen to refer to this process as “shipwrecking.” Receptor shipwrecking could be a valuable therapeutic approach; much in the same way as an antagonist precludes occupancy by an agonist, this approach would remove the receptor from the site at which it binds ligand or engages effector proteins. The ability to shipwreck GnRHRs, for example, might be a reasonable therapeutic approach to contraception; the shipwrecking of overexpressed receptors or enzymes might provide a means of treating cancer.
Receptor Rescue by Peptide Ligands
Although there is no documented case of pharmacological rescue by a peptide ligand, peptide ligands (e.g., GnRH) might be exploited to rescue receptors and increase surface expression. Although peptides on their own do not enter cells, cell entry can be mediated by receptor-regulated internalization, through the use of cell-penetrating peptides, or by stabilization of extant (but poorly expressed) plasma membrane proteins.
Conclusions
Recent data indicate that many loss-of-function mutations result from protein misrouting, rather than an intrinsic inability of the mutant receptor molecule to bind ligand or to interact with effectors. The surprising observation that this etiology is so common suggests a novel therapeutic approach that may be effective for a wide range of disorders. Likewise, it suggests an approach for increasing (or decreasing) the percentage of newly synthesized proteins in general that move from the endoplasmic reticulum to the plasma membrane.
Acknowledgments
Work in the authors’ laboratory is supported by NIH grants HD-19899, RR-00163, HD-18185 and TW/HD-00668.
Footnotes
-
Michael Conn is Associate Director of the Oregon National Primate Research Center, Special Assistant to the President of Oregon Health and Science University, and Professor of Physiology and Pharmacology at OHSU. He has written extensively on the molecular mechanism of hormone action and on topics related to neuroscience, endocrinology, physiology, pharmacology, and molecular biology. He is an outspoken supporter of the humane use of animals in research and the importance of public education about science.
-
Alfredo Leaños Miranda is a senior fellow supported by an international training program in reproduction sponsored by the Fogarty International Center. He presently practices medicine and has a research program at the Instituto Mexicano del Seguro Social in Mexico City.
-
Jo Ann Janovick is a senior research associate and has been in the Conn laboratory for 18 years, contributing to research, technique development, and training. She is a collector of Pacific Northwest Indian art and is devoted to her two dogs.
- © American Society for Pharmacology and Experimental Theraputics 2002

