Regulation of Gene Expression by the Hypoxia-Inducible Factors
Abstract
Many molecular and physiological responses to hypoxia in mammals are controlled by the transcription factors Hypoxia-Inducible Factor-1α (HIF-1α) and HIF-2α. Their ability to promote the transcription of hypoxia-inducible genes is mediated by protein stability and regulation of a C-terminal transactivation domain. Oxygen-dependent hydroxylation of conserved proline and asparagine residues in HIF-α are required for targeting HIF-α to proteasomes for destruction, and for inhibiting its capacity for CBP/p300-dependent transactivation, respectively. In hypoxia, the O2 required for prolyl and asparaginyl hydroxylation is limiting, and HIF-α is thus stabilized and competent for transcription. Because these proteins participate in angiogenesis, glycolysis, programmed cell death, cancer, and ischemia, HIF-α and its mediators are attractive therapeutic targets.
MRI slices of a hypoxic brain. Mike Smith and Bernie Dardzinski, Penn State University
INTRODUCTION
The term hypoxia describes a deficiency in normal oxygen delivery to cells, whether caused by an environmental reduction in oxygen supply, such as an increase in altitude, or a severe, localized ischemia caused by a disruption of blood flow to a given area. In general, responses to hypoxia in mammals can serve to stimulate the production of red blood cells through erythropoiesis (1), increase the supply of blood to affected areas by angiogenesis (2), or adapt cells to a greater reliance on glycolysis by increasing glucose transporter and glycolytic enzyme expression (3–5). The gene regulatory proteins Hypoxia-Inducible Factor-1α (HIF-1α) and HIF-2α mediate many of the molecular and physiological responses to hypoxia, and these factors play a crucial role in mediating the pathophysiology of numerous diseases, including cancer.
DISCOVERY OF HIF-1α AND HIF-2α
The gene for erythropoietin (EPO), a hormone that stimulates erythrocyte proliferation, undergoes hypoxia-induced transcription, and has been used as the prototype system in which to study hypoxia-responsive genes and transcription factors regulated by oxygen levels (1). The discovery of a hypoxia-inducible enhancer 3' to human EPO resulted in the demarcation of the hypoxia response element (HRE) required for hypoxia-mediated transcription (6). This permitted the subsequent isolation and cloning of the hypoxia-inducible dimer termed HIF-1 that binds to the HRE (7, 8), with the hypoxically inducible subunit referred to as HIF-1α.
HIF-1 DNA binding activity and HRE-mediated expression of a reporter gene are induced by hypoxia in both EPO- and non-EPO-producing cells (9–11), suggesting that HIF-1 can serve as an activator of genes other than EPO. Subsequently, several hypoxia-inducible genes have been found that have regulatory sequences with significant homology to the EPO 3' HRE, including the angiogenic mediator vascular endothelial growth factor (VEGF) (12), glucose transporter 1 (3), and glycolytic enzymes such as phosphoglycerate kinase and aldolase A (4, 5; see 13). Therefore, HIF-1α has a general role in multiple physiological responses to hypoxia, ranging from processes that quickly counteract oxygen deficiency (e.g., erythropoeisis and glycolysis), to long-term solutions such as angiogenesis. In addition, as the complement of target genes continues to expand, it appears that HIF-1α is involved in processes somewhat removed from these classical responses to hypoxia, including B lymphocyte development (14), carotid body function (15), adipogenesis (16), and apoptosis (17).
The closely related HIF-2α [also referred to as endothelial Per-ARNT-Sim (PAS) protein (EPAS), HIF-like factor (HLF), HIF-related factor (HRF) and member of the PAS superfamily 2 (MOP2) (18–21)] was identified and cloned shortly after the cloning of HIF-1α. RNA and protein analyses revealed that HIF-2α, but not HIF-1α, which is ubiquitously expressed, is found predominantly in the lung, endothelium, and carotid body (18, 19, 22). A number of structural and biochemical similarities are shared between HIF-1α and HIF-2α. However, there is evidence to suggest that their functions are not redundant and, although the complement of HIF-1α target genes has been extensively studied, only now is the physiological role of HIF-2α beginning to emerge.
HIF-1α AND HIF-2α ARE bHLH/PAS PROTEINS
HIF-1α and HIF-2α are members of the basic-Helix-Loop-Helix (bHLH)–Per-ARNT-Sim (bHLH–PAS) protein family (8, 18, 19) (Figure 1⇓). They form DNA binding heterodimers (8, 18, 19) with another bHLH–PAS protein called the aryl hydrocarbon receptor nuclear translocator [(ARNT), also termed HIF-1β], which was originally identified as the heterodimeric partner of the aryl hydrocarbon receptor (AHR) (23).
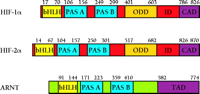
Schematic representation of human HIF-1a, HIF-2 a, and ARNT. HIF-1α, HIF-2α, and ARNT are basic-Helix-Loop-Helix–Per-ARNT-Sim (bHLH–PAS) proteins that contain an N-terminal bHLH domain and two PAS domains. HIF-1α and HIF-2α also contain an oxygen-dependent degradation domain (ODD) that mediates oxygen-regulated stability, and a C-terminal transactivation domain (CAD) whose transcriptional repression in normoxia is controlled by the inhibitory domain (ID). ARNT has a transactivation domain (TAD) that serves no function in the context of HIF-α activity. Amino acid numbers for each domain are indicated.
bHLH–PAS proteins contain an N-terminal bHLH domain and two PAS domains. The bHLH domain is required for dimerization and DNA binding, whereas the PAS regions appear to be involved in a variety of processes depending upon the protein (24). In the case of HIF-1α and HIF-2α, the functions of the PAS domains have not been be fully determined, although strong DNA binding activity of HIF-1α–ARNT and HIF-2α–ARNT heterodimers requires the presence of the PAS domains (25). The Drosophila melanogaster bHLH–PAS proteins Trachealess (Trh) and Single Minded (Sim) both heterodimerize with the Drosophila equivalent of ARNT and bind the same DNA sequence, but each heterodimer stimulates the expression of distinct target genes. Interestingly, a Trh-Sim chimera containing the Trh bHLH and the Sim PAS domain functions as a Sim protein (26). Therefore, the PAS domains (which do not bind DNA) confer the specificity of these two proteins towards target genes. Whether the PAS regions of HIF-1α and HIF-2α participate in specifying target gene selection in a fashion similar to that of Trh and Sim remains to be determined.
TRANSCRIPTION OF HYPOXIA-INDUCIBLE GENES BY HIF-1α AND HIF-2α
During hypoxia, HIF-1α and HIF-2α are translocated from the cytoplasm to the nucleus (27). Here they heterodimerize with the constitutively nuclear ARNT to enable binding to the cognate HRE DNA sequence and induce transcription of target genes (Figure 2⇓). All HIF-1α-inducible genes contain at least one copy of the core HRE consensus sequence 5' ACGTGC 3' (28), which is able to function regardless of location (11). Although HIF-2α can activate transcription of an HRE reporter gene (18, 19), distinct HIF-2α target genes have yet to be conclusively identified.
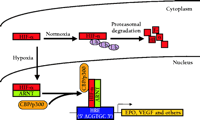
Hypoxia-inducible gene regulation by HIF-a. In normoxia, HIF-α is transcriptionally inactive and is rapidly degraded by the ubiquitin (Ub) proteasome pathway. In hypoxia, HIF-α undergoes protein stabilization and translocation from the cytoplasm to the nucleus, where it dimerizes with ARNT and associates with the transcriptional co-activators (as exemplified by CBP/p300) to induce the transcription of HRE-associated genes, such as erythropoietin (EPO) and vascular endothelial growth factor (VEGF). See text for details. CBP, cyclic AMP response element binding protein- (CREB)-binding protein; HRE, hypoxia response element.
STABILIZATION OF HIF-1α AND HIF-2α
ARNT protein is ubiquitously expressed in mammalian cells and is maintained at constant amounts regardless of oxygen partial pressures (29). In contrast, HIF-1α and HIF-2α are rapidly induced under hypoxic conditions and thus serve as the hypoxia inducible partners of their respective ARNT containing heterodimers.
Although the mechanisms by which HIF-1α and HIF-2α mRNA are regulated by hypoxia remain unclear, hypoxia-dependent transcription does not appear to play a major role in their induction (8, 29, 30). Rather, their expression is chiefly controlled post-translationally. Under conditions of normoxia, HIF-1α is virtually undetectable due to its rapid degradation through the ubiquitin-proteasome pathway (31–33), and has a short half-life (t1/2 ~ five minutes). However, when cells are exposed to hypoxia (i.e., 1% [O2]), its half-life is increased to approximately thirty minutes (32). Upon returning to normoxia, HIF-1α is once again unstable (8, 34). HIF-1α lability is mediated by the oxygen-dependent degradation domain (ODD) whose effects are suppressed by hypoxia (32). A similarly functioning ODD exists in HIF-2α (32, 35, 36). It is now clear that the ODD regulates the stability of HIF-1α and HIF-2α by interacting with the von Hippel-Lindau tumor suppressor gene product (pVHL).
INVOLVEMENT OF pVHL IN THE DEGRADATION OF HIF-1α AND HIF-2α IN NORMOXIA
Mutations in pVHL were originally described as the causative phenomena of an autosomal dominant cancer syndrome termed VHL disease, characterized by the development of multiple tumors, particularly clear-cell renal carcinomas, pheochromocytomas, and hemangioblastomas in the retina and central nervous system (37). pVHL functions as a component of a multimeric E3 ubiquitin ligase (38, 39) that includes elongins B and C, cullin 2, and Rbx1 (40–43). The entire E3 ubiquitin ligase complex displays much structural and functional similarity with the yeast SCF complexes, which consist of Skp1, Cdc53, cullin 2, Rbx1, and an F box protein, the latter of which functions similar to pVHL by aiding in substrate recognition (44). The mRNAs of hypoxia-inducible genes are aberrantly increased in cells defective for pVHL expression (45, 46), establishing a functional association between pVHL and HIF-α. By binding to HIF-1α and HIF-2α in normoxia, pVHL activates their oxygen-dependent degradation (47). In normoxic pVHL-defective renal carcinoma cells, HIF-1α and HIF-2α are active and stable; however, the expression of wild-type pVHL restores hypoxia-reversible lability to the proteins. In fact, a portion of the ODD of HIF-1α and HIF-2α (amino acids 549–572 and 517–534, respectively) is critical for binding to the β domain of pVHL and for mediating subsequent ubiquitination and proteasomal degradation (48–51). When a tumor grows to a critical size, oxygen can no longer be adequately delivered by diffusion. The subsequent intratumoral hypoxia activates HIF-α and induces the expression of genes (e.g., VEGF) that promote the formation of vessels to aid the tumor. One mechanism by which mutations in pVHL might lead to tumor formation is by permitting the stability and activity of HIF-α in normoxia and the subsequent expression of angiogenic factors even before the cancer’s exposure to a hypoxic stress.
Most but not all disease-causing pVHL mutations demonstrate a diminished ability to regulate HIF-α levels (52, 53), therefore the existence of other pVHL substrates and their role in tumor development cannot be ignored. For example, pVHL also binds to fibronectin and may be involved in extracellular matrix assembly (54). Mutated pVHL that was capable of regulating HIF-α had a diminished ability to associate with fibronectin (53). Thus, there may be a role in deregulated fibronectin in this as well as other VHL disease phenotypes. Identifying other proteins regulated by pVHL may help to explain tumor progression in patients where HIF-α regulation is normal, and to explain differences between types of VHL disease and the limited tissue origin of VHL disease tumors.
OXYGEN-DEPENDENT PROLINE HYDROXYLATION OF HIF-1α AND HIF-2α
Until recently, the mechanism by which reduced O2 in cells could translate to improved HIF-1α and HIF-2α function was poorly understood. A number of studies that demonstrated the association of HIF-α with the pVHL ubiquitination complex have now unexpectedly illuminated the nature of the cell’s oxygen sensors.
Hydroxylation of a proline within the ODD is responsible for the association of pVHL with HIF-1α or HIF-2α (55–57). pVHL binds directly to a short peptide derived from the ODD of HIF-1α only when a specific proline residue is hydroxylated (P564, corresponding to P531 in HIF-2α). Recent structural analysis of a P564-containing peptide derived from HIF-1α in a complex with pVHL and elongins B and C demonstrated the importance of the hydroxyproline in the interaction of pVHL with HIF-1α (58, 59).
Prolyl-4-hydroxylation was previously known to occur in collagen and proteins with collagen-like sequences (60). The characterized collagen prolyl-4-hydroxylases require O2, Fe2+, 2-oxoglutarate and ascorbate (60). Not surprisingly, binding of a HIF-1α-derived fragment (including P564) by pVHL—after treatment with whole cell extract believed to contain the hydroxylase activity—was enhanced by ascorbate, but was decreased by N-oxalylglycine (a competitive inhibitor of 2-oxoglutarate) or hypoxia (56). Furthermore, DMOG (dimethyl-oxalylglycine, a cell membrane permeant variant of N-oxalylglycine) increased the half-life of HIF-1α in cell culture (56), suggesting that a similar class of prolyl-4-hydroxylases modifies HIF-α. Most strikingly, this proposed O2-dependent hydroxylase was a candidate for the still hypothetical oxygen sensor.
HIF-α PROLYL-4-HYDROXYLASES
The collagen prolyl-4-hydroxylases consist of a tetramer of two α and two β units. Despite the functional similarities between HIF-α and collagen prolyl-4-hydroxylases, the pVHL binding domain of HIF-1α and HIF-2α contains no consensus sequences that resemble those in the collagen prolyl-4-hydroxylase substrates. Furthermore, human collagen prolyl-4-hydroxylases synthesized in insects were unable to hydroxylate HIF-1α peptides (55, 56). Indeed, the hypothesis that the HIF-α prolyl-4-hydroxylase was a new and distinct protein was subsequently confirmed by the discovery of the new class of HIF-α prolyl-4-hydroxylases (61, 62).
Epstein and coworkers analyzed HIF-1α regulation in Caenorhabditis elegans and subsequently identified the three mammalian HIF-α prolyl-4-hydroxylases (61). In C. elegans, the ortholog of HIF-1α was identified (see also 63) and shown to be stabilized in hypoxia and possess a pVHL-binding region similar to that in mammalian HIF-1α. In normoxia, this HIF ortholog interacted with pVHL only upon hydroxylation of a proline, similar to the finding for mammalian HIF-1α.
Epstein and coworkers subsequently identified putative HIF-α prolyl-4-hydroxylases by searching in C. elegans and mammalian databases for proteins that contained a conserved β-barrel jelly roll motif common to 2-oxoglutarate dependent oxygenases (as exemplified by the prolyl-4-hydroxylases of collagen). One such mammalian candidate is related to EGL-9 of C. elegans. A mutation in the egl-9 gene was first identified as causing an egg-laying abnormality, although no function had been attributed to the gene product (64). Three strains of worms carrying differing egl-9 mutations all had constitutively active HIF-1α that was not altered by oxygen tension (61). However, pVHL could interact with HIF-1α after the addition of recombinant wild-type EGL-9 without simultaneous treatment with worm extract (which would otherwise contain hydroxylase activity), and HPLC analysis demonstrated that the modification of HIF-1α was indeed prolyl-4-hydroxylation (61). Three human homologs of EGL-9 termed prolyl hydroxylase domain-containing (PHD) 1, 2, and 3 enhanced the capture of HIF-1α (and HIF-2α) by pVHL, and—at least in the case of PHD-1—appeared to be responsible for prolyl-4-hydroxylation of a HIF-1α-derived peptide containing P564 (61). Finally, a conserved LXXLAP sequence at the region of hydroxylation [determined upon the discovery of a second site of prolyl-4-hydroxylation in HIF-1α and HIF-2α (P402 and P405, respectively), (65)] was common to the HIF-α proteins of C. elegans and mammals, although their respective hydroxylases were not cross-reactive (61).
In another study, Bruick and McKnight searched the GenBank database for sequences related to the α subunit of the collagen prolyl-4-hydroxylases (the catalytic component of the multimer). Of five candidates, one displayed HIF-α hydroxylase activity [designated HIF prolyl hydroxylase 1 (HPH-1)] and this protein in turn had two highly similarly sequenced and functional paralogs (HPH-2 and HPH-3). The Drosophila melanogaster HPH homolog was identified, and reduced amounts of HPH mRNA in insect cells—as mediated by RNA interference—corresponded to an increase in normoxic levels of Sim a [Similar a, a Drosophila HIF-1α homolog—not to be mistaken with the previously mentioned Single Minded (Sim)] (62).
To summarize, the three HIF-α prolyl-4-hydroxylases function to target HIF-1α and HIF-2α for binding to pVHL and, ultimately, degradation by the ubiquitin proteasome pathway (Figure 3⇓). Furthermore, these proteins were found to catalyze a previously unknown enzymatic function and, unlike the collagen prolyl-4-hydroxylases that are composed of two α and two β subunits, consist only of the catalytic α unit. In other respects, these hydroxylases share much in common, such as their dependence on Fe2+ and 2-oxoglutarate and, most importantly, in the context of HIF-α regulation, dioxygen. The absolute requirement of O2 for hydroxylase activity suggests that these proteins may serve as the primary oxygen sensor in cells and might also explain the exponential increase in HIF-1α protein levels as cells are exposed to a decreasing partial pressure of O2 (66).
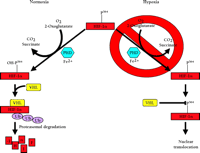
Oxygen dependent degradation of HIF-1a and HIF-2a. In normoxia, a specific proline within HIF-1α (P564, or P531 in HIF-2α) is hydroxylated by O2-, 2-oxoglutarate-, ascorbate-, and Fe2+-dependent HIF-α prolyl-4-hydroxylases (PHDs). This targets the HIF-α protein for association with the von Hippel-Lindau protein (pVHL) that serves as the HIF-α recognition component of an E3 ubiquitin ligase complex (VHL) also containing elongins B and C, cullin 2, and Rbx 1 and results in degradation of HIF-α protein via the ubiquitin proteasome pathway. In hypoxia, the required O2 for prolyl-4-hydroxylase activity is deficient. As a result, HIF-α protein is not degraded because the prolyl-4-hydroxylation required for targeting by the VHL complex is inhibited.
TRANSCRIPTIONAL ACTIVATION OF HIF-1α AND HIF-2α
Despite the crucial role of prolyl-4-hydroxylation and pVHL association in regulating HIF-1α and HIF-2α protein levels, their stabilization alone is not sufficient to permit their full transcriptional activation. HIF-α contains two hypoxia-inducible transactivation domains separated by an inhibitory domain (ID) that inhibits transactivation domain function in normoxia (67). One transactivation domain (the N-terminal transactivation domain or NAD) is encompassed within the ODD of HIF-α. Thus, transcriptional activity appears to be directly correlated with protein stability. The dominant transactivation domain consists of the C-terminal forty amino acids (CAD). When expressed as an isolated protein domain, CAD is constitutively active, but in the presence of the adjacent ID, CAD activity is repressed at normoxia and induced by hypoxia independent of protein stability (35, 61). Recent studies have now shed light on how the ID regulates CAD activity in HIF-1α and HIF-2α, and also confirm the two-step model of HIF-α regulation. Thus, hypoxia serves to promote both protein stabilization and enhance transcriptional activity by distinct but related molecular mechanisms.
OXYGEN-DEPENDENT ASPARAGINE HYDROXYLATION OF HIF-1α AND HIF-2α
When fused to the Gal4 DNA binding domain (Gal4DBD), the final 100 amino acids of HIF-1α and HIF-2α (the CAD plus the C terminus of the ID) cause a substantial increase in Gal4 response element-(GRE)-mediated reporter activity in hypoxia without altering protein stability (31, 67). Lando and coworkers established stable mammalian cell lines expressing the final 100 amino acids of HIF-1α or of HIF-2α, exposed the latter line to normoxia and hypoxia, purified the protein and searched for posttranslational modifications using mass spectrometry (68). The truncated HIF-2α purified from normoxic cells contained a specific hydroxylated asparagine (N851) that was essentially unmodified in protein purified from hypoxic cells. Because this asparagine is conserved in HIF-1α (N803), it was hypothesized that the failure to hydroxylate this amino acid was the mechanism by which hypoxia modulates CAD activity. Furthermore, it suggested that the enzymes that target HIF-α for normoxic degradation were different from those that inhibit assembly of the transcriptional machinery by the CAD.
Asparaginyl–aspartyl hydroxylases have been identified that catalyze the hydroxylation of specific asparagine or aspartic acid residues of epidermal growth factor-like domains and function on the consensus sequence CXN/DXXXXP/YXCXC (69, 70). Like the HIF-α prolyl-4-hydroxylases, they are dependent on 2-oxoglutarate, Fe2+, and O2. Treatment of cells with the 2-oxoglutarate inhibitor DMOG activated Gal4DBD/HIF-1α CAD and Gal4DBD/HIF-2α CAD to an extent comparable with hypoxia, whereas mutation of the crucial asparagine in HIF-1α and HIF-2α resulted in maximum transcriptional activity that was not enhanced by hypoxia or DMOG (68), confirming the importance of this residue in regulating CAD activity. In the context of the full-length protein, full transcriptional activity of HIF-1α and HIF-2α in normoxia could be obtained only through mutation of both the critical proline in the ODD and the asparagine in the CAD. A mutation only in the ODD creates a constitutively stable protein with low transcriptional activity, whereas if only the CAD is mutated, the protein is labile, although presumably active (68).
Asparaginyl hydroxylation of the CAD inhibited its recruitment of the CH1 domain of the transcriptional coactivator cyclic AMP response element-binding protein-(CREB)-binding protein (CBP), suggesting that asparaginyl hydroxylation in normoxia inhibits the previously observed hypoxic recruitment of CBP/p300 and other coactivators such as SRC-1 (27, 36, 71–74). In fact, structural analysis of the HIF-1α CAD, when associated with CBP/p300, demonstrated that N803 is deeply buried within the complex, and it is postulated that hydroxylation of this asparagine would render the recruitment of CBP/p300 by the HIF-1α CAD unlikely (75, 76).
Thus, there are two major steps required for the hypoxic induction of HIF-1α and HIF-2α function: i) inhibition of the oxygen-dependent prolyl-4-hydroxylation in the ODD to prevent the association with the pVHL–ubiquitin-ligase complex, and subsequent destruction of HIF-α by the ubiquitin proteasome pathway; and ii) inhibition of the oxygen dependent asparaginyl hydroxylation in the CAD to permit association with CBP/p300 and assembly of the transcriptional co-activator complex.
HIF-α ASPARAGINYL HYDROXYLASES
Hydroxylation of substrates by known asparaginyl–aspartyl hydroxylases has yet to be linked to biological function, although studies involving hydroxylase-deficient mice seem to indicate roles for these proteins in tumor suppression and in facial and limb development (77).
The first protein shown to cause inhibition of CAD function in normoxia was factor inhibiting HIF-1 (FIH-1), which was identified by a yeast two-hybrid screen using HIF-1α 576–826 as bait (78). FIH-1 reduces the activity of cotransfected HIF-1α, and forms a ternary complex with pVHL and HIF-1α in vitro (78). This suggests that prolyl-4-hydroxylation of the conserved proline within the ODD permits the formation of the HIF-α–pVHL complex, which recruits FIH-1 to the CAD for transcriptional inhibition, thus indirectly coupling protein instability and CAD inhibition in normoxia.
However, there is evidence to separate pVHL from involvement with reduced CAD activity in normoxia. First, the hydroxylation-dependent regulation of the CAD appears to be independent of pVHL; a Gal4DBD–HIF-1α 740–826 chimera displays similar amounts of hypoxia-inducible reporter expression in the presence or absence of pVHL (79). There is also evidence to suggest that HIF-α asparaginyl hydroxylase activity is pVHL-independent. Specifically, purified CADs (consisting of only the C-terminal 100 amino-acids of HIF-1α and HIF-2α, and thus no pVHL binding site) contained a hydroxylated asparagine under normoxic, but not hypoxic, conditions (68). More importantly, it is now known that FIH-1 is a 2-oxoglutarate-, Fe2+-, and O2-dependent asparaginyl hydroxylase that hydroxylates N803 and N851 of HIF-1α and HIF-2α, respectively, and inhibits their association with the CH1 domain of CBP (80, 81).
Initially, FIH-1–mediated repression of the HIF-α CADs could be ablated with DMOG treatment, suggesting that FIH-1 may be a 2-oxoglutarate-dependent hydroxylase (80). Mass spectrometry of recombinant HIF-2α 774–874 treated with FIH-1, Fe2+, ascorbate, and 2-oxoglutarate resulted in complete hydroxylation of N851, although this residue was unmodified when the inhibitor DMOG was included (80). Furthermore, FIH-1 inhibited the interaction between 35S-labelled HIF-2α 774–874 and the CH1 domain of CBP unless DMOG had been added, or N851 had been mutated (80). These results occurred in the absence of pVHL, confirming that FIH-1 is a 2-oxoglutarate-, O2-, and Fe2+-dependent enzyme that hydroxylates a conserved asparagine in the CAD of HIF-α, and inhibits the association of HIF-α with CBP/p300 and the subsequent assembly of the transcriptional co-activator complex. Presumably, hypoxia blocks this inhibition by depriving the enzyme of the required O2, ultimately permitting the potentiation of HIF-α CAD activity (Figure 4⇓). Although HIF-α prolyl-4-hydroxylase-mediated inhibition of HIF-α activity is reduced in hypoxia (62), FIH-1 can inhibit Gal4DBD–HIF-1α 727–826 in both normoxia and hypoxia when overexpressed in cell culture (80), so the possibility that the functions of FIH-1 and also the prolyl hydroxylases are mediated by factors in addition to dioxygen affinity cannot be discounted. For example, FIH-1 might be able to compete with CBP/p300 for binding to the HIF-α CADs even in hypoxia.
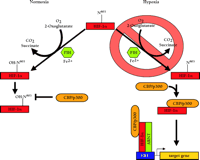
Oxygen-dependent transcriptional inactivation of HIF-1a and HIF-2a. In normoxia, a specific asparagine within HIF-1α (N803, or N851 in HIF-2α) is hydroxylated by an O2-, 2-oxoglutarate-, and Fe2+-dependent HIF-α asparagine hydroxylase (FIH-1). This inhibits the direct interaction of the C-terminal transactivation domain (CAD) with CBP/p300 and, ultimately, the assembly of the transcriptional coactivator complex. In hypoxia, the required O2 tension for asparaginyl hydroxylase activity is deficient. As a result, CBP/p300 can associate with the unhydroxylated CAD and potentiate transcriptional activtity.
Thus, the discovery of the HIF-α prolyl-4-hydroxylases and asparaginyl hydroxylases and their respective independent involvement in regulating protein stability and transcriptional activity supports the two-step model of HIF-α function (Figure 5⇓). Future studies will presumably concentrate on characterizing the oxygen binding constants of each of the enzymes, their expression patterns, and specific roles in vivo in regulating HIF-α activity. It will also be intriguing to identify other substrates of the HIF-α prolyl-4-hydroxylases and aspartyl–asparaginyl hydroxylases and to determine their fates after hydroxylation. These could be proteins that are controlled posttranslationally to aid adaptation to hypoxia. Hydroxylation could even confer novel functional switches to these substrates.
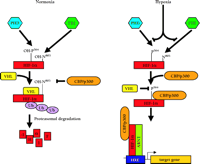
Two step model for the induction of HIF-1a and HIF-2a function. There are two major steps required for the hypoxic induction of HIF-1α and HIF-2α activity: i) inhibition of the oxygen-dependent prolyl-4-hydroxylation in the ODD to prevent the association with the pVHL–E3-ubiquitin-ligase complex (VHL), and ii) subsequent destruction by the ubiquitin (Ub) proteasome pathway, and inhibition of the oxygen-dependent asparaginyl hydroxylation in the CAD to permit association with CBP/p300, subsequent assembly of the transcriptional co-activator complex, and transcription of HIF-α target genes. The prolyl-4-hydroxylases (PHDs) and asparaginyl hydroxlase (FIH-1) function independently, with HIF-α currently as their only known substrate.
OTHER FORMS OF HIF-1α AND HIF-2α REGULATION
Although it is now clear that both classes of hydroxylases are vital oxygen sensors in HIF-α regulation, it would be premature to suggest that the HIF-α prolyl-4-hydroxylases and asparaginyl hydroxylases are the only oxygen sensors and mediators of HIF-1α and HIF-2α stability and transcriptional activity.
An array of cytokines including insulin and the IGFs has been shown to regulate HIF-α through the phosphatidylinositol-3' kinase (PI3K) pathway, probably through an increased rate of translation (summarized in 82). There is also a proposed role for reactive oxygen species (ROS) in the control of HIF-α, although it is unclear whether they induce (83) or repress (34) HIF-α function.
HIF-1α interacts with the p53 tumor suppressor gene in vivo (84), and promotes the ubiquitin proteasome-mediated destruction of HIF-1α through the Mdm-2 E3 ubiquitin ligase (85). Whereas pVHL performs a role in normoxia by promoting the degradation of the appropriately prolyl-4-hydroxylated HIF-α subunit, p53 might regulate HIF-α concentrations in hypoxia. Indeed, this is particularly likely given that angiogenesis of tumors derived from p53−/− lines is greater than in p53+/+ lines and more specifically, p53−/− lines showed greater HIF-1α-mediated VEGF activity in hypoxia than do p53+/+ counterparts (85). Elucidating where p53 interacts with HIF-1α and HIF-2α and what modifications, if any, permit or inhibit association may shed light on p53’s role in controlling HIF-α function.
DIFFERENTIAL FUNCTIONS OF HIF-1α AND HIF-2α
HIF-1α and HIF-2α share 48% amino acid identity (19), and both are stabilized and transcriptionally activated by hypoxia; however, there is strong evidence to suggest each protein possesses unique qualities and distinct biological roles, and although many direct HIF-1α target genes have been widely studied (listed in 13), none has been positively identified for HIF-2α.
This is particularly evident in light of studies on HIF-1α−/− or HIF-2α−/− mice. Hif1α−/− mouse embryos die by E11 as a consequence of defective vascularization as well as heart malformation and incomplete neural tube closure (86, 87). Furthermore, in Hif1α−/− embryonic stem (ES) cells exposed to 20% O2 (normoxia), the expression of mRNA transcripts encoding glucose transporters and glycolytic enzymes, and the rate of cell division are decreased compared to those of wild-type ES cells (86). When exposed to 1% O2 (hypoxia), the expression of VEGF mRNA is lower than that found in similarly treated wild-type cells (86). In contrast, mice embryos lacking HIF-2α develop a normal circulatory system; however, they die by E16.5 as a result of blood congestion caused by bradycardia (22). A role for HIF-2α in the biosynthesis or release of the heart-rate–mediating catecholamines (such as norepinephrine) has been hypothesized given that HIF-2α is highly expressed in the organ of Zuckerkandl (the primary source of embryonic catecholamine production) and that HIF-2α-deficient embryos contain a significantly reduced level of catecholamines. Embryonic lethality was rescued by the addition of DOPS (a precursor to norepinephrine) to the mother’s diet, although the mice died within twenty-four hours of birth, because of the discontinued supply of DOPS (22). However, a second study implicates HIF-2α in vascular remodelling (88). In this study, Hif2α−/− mice died by E12.5. Although vascularization of the embryo had taken place, there was inadequate fusion or subsequent vascular assembly. Recently, a third report has associated HIF-2α loss in mice with an impediment in fetal lung maturation (89). Those Hif2α-/- mice that did not die from cardiac failure before E13.5 died shortly after birth due to respiratory distress syndrome caused by decreased VEGF expression in alveolar epithelia, resulting in the insufficient production of lung surfactant. The difference in phenotypes, and subsequently, the postulated roles for HIF-2α in the three studies are unclear, but they are thought to be a consequence of utilizing different ES cell and mouse strains. Nonetheless, ubiquitously expressed HIF-1α does not compensate for the lack of HIF-2α in any of the reports, and thus HIF-2α appears to perform a distinct role in embryogenesis and may do also in the developed organism.
The DNA binding activity of HIF-2α, but not HIF-1α, is regulated by reducing conditions in the cell. A cysteine residue present in the basic DNA binding region of HIF-2α must be reduced by the redox factor Ref-1 before HIF-2α can bind DNA (90). This is not the case for HIF-1α, where serine is present in the equivalent position (the only difference between the amino acid sequences of the basic regions in their DNA binding regions) and DNA binding is constitutive (90). This identifies at least one mechanism of differential regulation of HIF-1α and HIF-2α activity.
The renal cell carcinoma cell line 786-0 produces detectable levels of HIF-2α, but not HIF-1α. Because 786-0 expresses a non-functioning, truncated pVHL, HIF-2α expression is not regulated by hypoxia (47). Recent studies using the 786-0 line have helped resolve HIF-2α’s function in vivo and demonstrated that pVHL’s function as a tumor suppressor protein is linked, at least in part, to its ability to regulate HIF-2α expression (91, 92). Expression of HIF-1α mutated at P564 in 786-0 cells where wild-type pVHL had been stably reintroduced was sufficient to recover the morphological phenotype observed in the originally pVHL-deficient cells. However, unlike constitutive HIF-2α expression, the deregulated expression of the P564-containing HIF-1α mutant could not increase the rate of tumor growth when these cells were injected into mice (91), suggesting a tumor-promoting role for HIF-2α, and not HIF-1α, in the context of 786-0 cells. Although all of these studies provide clues as to the different roles of HIF-1α and HIF-2α, a great deal more work is required to understand the mechanisms of differential activation, specific target genes, and the respective pre- and post-natal roles for HIF-1α and HIF-2α.
THE ROLE OF HIF-1α AND HIF-2α IN DISEASE
VHL disease now appears to be manifested in most circumstances as a consequence of deregulated HIF-α, which is involved in tumor progression. However, it was evident prior to the appreciation of pVHL’s function that HIF-α plays a role in non-VHL-associated cancers. As it grows, a tumor ultimately requires neovascularization in order to supply its cells with oxygen, and to overcome the deficiencies in gas (O2) diffusion. Hypoxic regions within the tumor can increase HIF-α stability and transcriptional activity, VEGF expression, and ultimately, angiogenesis; HIF-1α and HIF-2α are overexpressed in many tumors, presumably as a result of hypoxia within the tumor (reviewed in 82). Indeed, early studies demonstrated not only the hypoxia inducible nature of VEGF expression in cell culture, but also its increased expression in vascularizing tumors (2, 93). The subsequent identification of VEGF as a HIF-1α target implicated the latter in tumor angiogenesis (94). This was confirmed with the use of HIF-1α−/− ES cells (87), which displayed a reduced ability (as compared with wild-type cells) to form tumors in immunocompromised mice because of reduced size and decreased vascularization, presumably due to compromised VEGF expression (87, 95). Interestingly, hypoxia-induced apoptosis in HIF-1α+/+ ES cells was increased in comparison to that in HIF-1α−/− cells, suggesting that HIF-1α may play a role in inhibiting cell growth in some tumors while promoting growth in others (95). HIF-1α has also been implicated in the Warburg effect, a poorly understood phenomenon where cancer cells display a high rate of aerobic glycolysis (96). It has been proposed that the observed increase in glycolytic enzyme expression is caused by constitutive HIF-1α activity (97). Conversely, it has also been suggested that the glycolytic end products lactate and pyruvate may induce HIF-1α accumulation (98). Hence, in addition to an important role in tumor angiogenesis, HIF-α is also implicated in regulating other major biochemical processes involved in tumor formation and progression.
The development of a potential antiangiogenic therapy has stemmed from studies using a peptide derived from the last forty-one amino acids of HIF-1α—essentially the CAD—that is able to bind and sequester the CH1 domain of CBP (99). This peptide attenuated hypoxia-induced reporter activity in cell culture and reduced VEGF mRNA expression in normoxia and hypoxia (99). Furthermore, expression of this peptide in tumor cell lines reduced tumor growth in mouse models with diminished vessel density. This suggests that the HIF-α–CBP interaction may be a good therapeutic target (99). However, target specificity is a concern because the CH1 domain associates with numerous transcription factors, and any inhibitor may serve as a negative regulator of these proteins as well, as was the case with the transcription factor STAT2 in this study (99).
The inhibitory PAS protein (IPAS) consists of a bHLH and a PAS domain but no transactivation domain (100). In vitro binding studies demonstrate that IPAS binds to HIF-1α (but not ARNT), and the resultant heterodimer does not associate with an HRE, suggesting that IPAS may serve as a dominant negative regulator of HIF-1α (100). IPAS mRNA is expressed predominantly in the eye, suggesting a role in inhibiting angiogenesis in the cornea (100). Indeed, not only do tumors derived from cells that express IPAS display a slower growth rate and decreased vascular density when compared to those initiated from wild-type cells, but primary corneal cells transfected with an antisense IPAS expression vector displayed enhanced hypoxic inducibility of VEGF mRNA (100). Furthermore, expression of antisense IPAS in the cornea of mice induced angiogenesis of the cornea (100). It is imperative that the cornea experiences little vascularization because the presence of vessels would severely inhibit the passage of light to the retina. The identification of IPAS primarily in the eye suggests a mechanism by which HIF-1α-induced angiogenesis is reduced in periods of localized hypoxia, such as during closure of the eyelids. Interestingly, IPAS, identified by Gu and coworkers, is a splice variant of the HIF-3α paralog (101, 102). Accounting for this, it is odd that IPAS associates with HIF-1α and not ARNT (100), although this would seem preferable, given the involvement of ARNT in the function of other transcription factors. Nonetheless, if the IPAS binding capacity is limited to interactions with HIF-α, IPAS or a similarly functioning variant could be administered as an antiangiogenic agent in newly established tumors.
Increased HIF-α expression also holds great promise for the treatment of certain diseases. Strokes and heart attacks are caused by localized hypoxia manifested as cerebral and myocardial ischemia, respectively. A potential preventive therapy for these conditions might involve HIF-1α- or HIF-2α-activated VEGF expression to induce neovascularization of the target area (103), which would provide an increased blood flow and oxygen supply to disease-prone areas of the brain and heart and reduce harmful responses to ischemia. In fact, recent studies have shown that overexpressing HIF-1α in mice promotes enhanced vascularization with few of the defects seen when VEGF alone is overexpressed, such as vascular leakage and inflammation (104). Therefore, the administration of drugs that activate HIF-1α, or HIF-2α, or both, and induce VEGF-controlled vascularization of the effected area is a viable option in the treatment of ischemic diseases. The recent discovery of hydroxylases as key oxygen sensors make them attractive targets because disruption of their activity should collectively stabilize and activate HIF-α thereby inducing angiogenesis. The importance of two classes of hydroxylases and multiple genes also suggests that specific targeting may be able to achieve differential results.
CONCLUSION
There has been a rapid advance in the understanding of the molecular mechanisms that regulate responses to hypoxia in mammalian cells. The most recent studies have finally unravelled how a reduction in cellular oxygen can be sensed and translated into enhancing the activity of HIF-1α and HIF-2α, and ultimately, counteracting oxygen deprivation. Given their clear importance to tumor vascularization and ischemic disease, HIF-1α and HIF-2α or any of the hydroxylases that mediate their function should serve as effective therapeutic targets.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Daniel J. Peet, PhD, (left) is the W. Bruce Hall Research Fellow in the Department of Molecular Biosciences at the University of Adelaide, is a member of the Centre for the Molecular Genetics of Development, and is a member of the Whitelaw Laboratory. Address comments to DJP. E-mail daniel.peet{at}adelaide.edu.auMurray L. Whitelaw, PhD, (middle) is a senior lecturer in the Department of Molecular Biosciences (Biochemistry) at the University of Adelaide and is a member of the Centre for the Molecular Genetics of Development. Anthony O. Fedele (right) is a postgraduate student in the Whitelaw Laboratory working toward his doctorate.

