Adrenoceptors: A Convergence of Thinking
- Martin C. Michel, MD1,
- Jeffrey R. Jasper, PhD2 and
- Paul Insel, MD3
- 1completed his doctoral work at the University of Essen in Germany and postdoctoral training at UCSD and the University of Essen. He is currently Professor of Internal Medicine at the University of Essen.
- 2completed doctoral work in Pharmacology at UCSD, postdoctoral training at Stanford University, and worked at Merck Research Laboratories and Roche Bioscience before joining Theravance, Inc., where he is currently Director of Biochemistry.
- 3graduated from the University of Michigan, pursued postgraduate training at Harvard University, the NIH, and UCSF, and held a faculty position at UCSF before moving to the Departments of Pharmacology and Medicine at UCSD, where he is Professor and Director of the Medical Scientist Training Program.

On July 12-14, 2002, the Sixth IUPHAR Satellite on Adrenoceptors and the Second Annual ASPET GPCR Symposium were jointly held
in Rohnert Park (CA, USA). Meeting organizers: D. Bylund, B. Kobilka, S. Lanier, and R. Ruffolo. Meeting Report by M. Michel,
J. Jasper, and P. Insel.
Abstract of posters presented at the meeting are published in Receptors & Channels 8: 123-34, 2002.
Adrenoceptors regulate a large number of physiological responses, and do so by undergoing activation by norepinephrine (noradrenaline) or epinephrine (adrenaline); a large variety of additional adrenoceptor agonists and antagonists are used experimentally and clinically. Moreover, as prototypical G protein–coupled receptors (GPCRs), adrenoceptors interact with members of all four of the major classes of heterotrimeric G proteins (i.e., Gs, Gi, Gq/11, and G12/13) and thus serve as important model systems to explore activation and deactivation (desensitization) of G protein–signaling pathways.
Whereas past IUPHAR Satellite Meetings on adrenoceptors emphasized adrenergic physiology and pharmacology, and tracked developments in cloning of receptors, the main scientific motif of the 2002 Satellite proved to be one of convergence. This theme began with a review of past IUPHAR adrenoceptor satellites (E. Szabadi, University of Nottingham, UK), and emphasized the interplay of experimental and clinical aspects of adrenoceptor pharmacology. The use of cellular and molecular biological techniques dominated the presentations. Here, we divide the Satellite presentations into four categories: structure and function; trafficking and compartmentation; knockout animals and novel pharmacological agents; and the genetics of mice and men.
STRUCTURE AND FUNCTION
Although sensitivity to prazosin and propranolol are the hallmarks of α1- and β-adrenoceptors, respectively, prazosin-insensitive states of the α1A-adrenoceptor and propranolol-insensitive states of the β1-adrenoceptor have been identified and termed “β1L” and “β4,” respectively. However, the structural basis, indeed the definitive existence of such distinct alternative forms, remains elusive (P. Hieble, Glaxo SmithKline, King of Prussia, PA, USA). Of note, extension of adrenoceptor cloning to the zebrafish (Danio rerio) has revealed the existence of additional α2-adrenoceptor subtypes not found in mammals (J. Ruuskanen, University of Turku, Finland). Thus, the total number of mammalian adrenoceptor subtypes (α1A/B/D, α2A/B/C, and β1/2/3) remains at nine, although splice variants have been identified for certain adrenoceptor subtypes, such as β3-adrenoceptors (R. Summers, Monash University, Melbourne, Australia). The precise physiological role of such splice variants remains ill defined.
Consistent with the theme of convergence, M. Bouvier (Université de Montreal, Canada) presented a comprehensive discussion of receptor dimerization. Early studies on the dimerization of GPCRs, and adrenoceptors in particular, relied primarily on co-immunoprecipitation and overexpression in heterologous systems. Bioluminescence-resonance energy transfer (BRET), used by Bouvier and colleagues, now suggests that dimerization is the rule rather than the exception in GPCR biology and can involve formation of both homo- and heterodimers. Newer BRET techniques reveal that receptor dimerization occurs in the endoplasmic reticulum prior to receptor localization to the plasma membrane. Both agonists and antagonists can alter the BRET-defined patterns of dimerization, but such pattern changes appear to result primarily from conformational changes of existing dimers, rather than from dimerization de novo. Whereas Bouvier focussed on β-adrenoceptors, K. Minneman (Emory University, Atlanta, GA, USA) discussed the possible oligomerization of α1-adrenoceptors, which appear to be detected by immunoprecipitation but not by photoaffinity labeling.
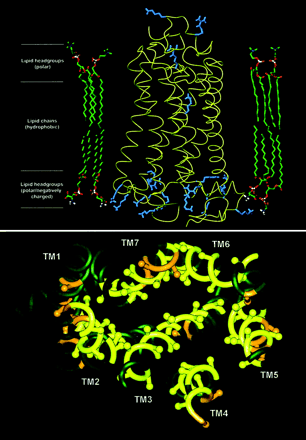
Adrenoceptors are all members of the G protein–coupled receptor (GPCR) superfamily, for which the crystal structure of one member, rhodopsin, has been solved. Schematics of the rhodopsin peptide and its 7-transmembrane (TM) domains are shown. As demonstrated at a recent IUPHAR satellite/ASPET GPCR symposium, convergence of adrenoceptors may occur with each other (oligomerization), with other proteins, and in signaling microdomains. In addition, molecular insights into adrenoceptors are converging with whole animal and clinical perspectives on catecholamine pharmacology.
Earlier concepts of adrenoceptor function emphasized the ternary complex of receptor, G-protein, and effector, but additional partnerships between adrenoceptors and accessory proteins are now recognized. These interactions occur between: phosphorylated (and thereby activated) receptors with specific domains on arrestins (V. Gurevich, Vanderbilt University, TN, USA), as well as between receptors and RGS (regulator of G-protein signaling) proteins (M. Ishii, Osaka University, Japan). B. Zheng (University of California, San Diego, USA) reported the identification of RGS-PX1, the previously elusive RGS for the Gs protein. RGS-PX1 increases the GTPase activity of Gs and thereby inhibits β-adrenoceptor function. In addition, the concomitant stimulation of several G-proteins may be necessary to elicit downstream responses, such as in the activation of c-Jun N-terminal kinase by cardiac α1-adrenoceptors (H. Kurose, Kyushu University, Japan).
TRAFFICKING AND COMPARTMENTATION
Another form of convergence involves the co-localization of proteins on the plasma membrane or in intracellular compartments. A considerable fraction of certain adrenoceptors occurs in intracellular compartments that can be visualized using fluorescent protein–tagged receptors both in transfected cells and in transgenic animals (J. McGrath, Institute of Biomedical and Life Science, Glasgow, UK). M. von Zastrow (University of California, San Francisco, USA) used the β2-adrenoceptor and the δ-opioid receptor to exemplify that receptors can undergo different fates upon agonist-induced internalisation: whereas the β2-adrenoceptor is primarily recycled to the cell surface, the δ-opioid receptor is directed towards degradative events in lysosomes. This differential sorting seems to be governed by sequences in the C terminus of each receptor.
Recent data have emphasized the co-localization of certain adrenoceptors with other signaling molecules in caveolae, membrane microdomains enriched in cholesterol, sphingolipids, and the protein caveolin (B. Kobilka, Stanford University, CA, USA; S. Steinberg, Columbia University, NY, USA). Multiple techniques have been used to demonstrate microdomain-dependent signaling efficacy; accumulating evidence suggests that differences in the signaling of cardiac β1- and β2-adrenoceptors are at least partly due to their differential association with other signaling proteins in caveolae.
KNOCKOUT ANIMALS AND NOVEL PHARMACOLOGICAL AGENTS
The convergence of modern molecular techniques and more classical pharmacological approaches was demonstrated by the use of knockout mice to assess the role of particular adrenoceptors. Mice lacking each of the nine cloned adrenoceptor subtypes have been generated. Whereas α1A-adrenoceptor knockout mice exhibit lowered basal blood pressure (P. Simpson, University of California, San Francisco, USA), α1B-adrenoceptor knockout mice do not show a change in basal blood pressure, but instead demonstrate several behavioral phenotypes (S. Cotecchia, Université de Lausanne, Switzerland). Because the α1D-adrenoceptor protein is only sparsely expressed in mice, α1A/B double knockout mice effectively lack a1-adrenoceptors, especially in peripheral tissues. Simpson demonstrated that such mice exhibit normal blood pressure but decreased cardiac size and output, suggesting a decrease in peripheral resistance; this phenotype only appeared in males. Kidneys were also larger in the double knockout females, but not in males.
Among the α2-adrenoceptor subtype knockout mice, the most pronounced phenotypes arise from the knockout of α2A-adrenoceptors (M. Scheinin, Turku University, Finland). Heterozygous knockout mice (i.e., retaining only one parental copy of the α2A-receptor–encoding gene) lose the sedative but not the antihypertensive response to the α2-adrenergic agonist moxonidine (C. Tan, Vanderbilt University, TN, USA). Consistent with a dominant role of α2A-adrenoceptors relative to α2B-adrenoceptors, highly selective α2B-adrenoceptor agonists have recently been found to have minimal in vivo effects, other than reducing intraocular pressure, in experimental animals (D. Gil, Allergan, Irvine, CA, USA). Subtype-selective, peripherally acting α2-adrenoceptor antagonists may have beneficial effects on adipocytes in which receptor activation has an anti-lipolytic activity, thereby promoting fat storage (M. Lafontan, Institut Louis Bugnard, Toulouse, France).
B. Kobilka used cardiac myocytes from β1-, β2-, and β1/β2-double knockout mice to illustrate specific cellular domains that constrain downstream signaling. Moreover, he showed that adding back individual receptor subtypes to the double knockouts can provide useful information regarding receptor compartmentation and signaling.
Alterations of the physiological stoichiometry of elements in receptor/G protein/effector cascades may play important roles in receptor signaling, because some components of the pathway may be more rate-determining than others (P. Insel, University of California, San Diego, USA). In this regard, mice lacking type V adenylyl cyclase, a main Gi-regulated isoform of cardiac adenylyl cyclase, exhibit diminished cardiac responses to β-adrenoceptor stimulation and lose response to muscarinic receptor stimulation (C. Homcy, Millenium Pharmaceuticals, Cambridge, MA, USA).
GENETICS OF MICE AND MEN
Complementing and extending the genetic strategies in murine systems are the evolving data regarding polymorphisms and genetic variants of adrenoceptors in humans. For example, a variant in the α2B-adrenoceptor–encoding gene is associated with an increased incidence of myocardial infarction; this variant is enriched in Finnish patients who have died suddenly (M. Scheinin, University of Turku, Turku, Finland). The α2C-adrenoceptor subtype, involved in prejunctional inhibition at low stimulation frequency, occurs as a variant (deletion of amino acids 322–325) that is more frequent in African American than Caucasian subjects; in the former group this variant is associated with a fivefold greater risk of developing congestive heart failure, and this effect may be additive or perhaps even synergistic with expression of an Arg389 (vs Gly389) variant of the β1-adrenoceptor (S. Liggett, University of Cincinnati, OH, USA). β-Adrenoceptor polymorphisms, in particular certain β2-adrenoceptor polymorphisms, may also influence the course of acquired and monogenic disorders, and may also affect responses of humans to β-adrenergic agonists in vivo (P. Insel).
SUMMARY AND PERSPECTIVE
Given the convergence of themes presented at the Satellite, it was fitting that L. Limbird (Vanderbilt University, Nashville, TN, USA) presented a well-received synthesis of past history, present work, and potential future developments. Limbird nicely summarized and integrated classical experiments in pharmacology and physiology with evolving approaches in cell and molecular biology. The meeting thus demonstrated the power that the convergence of approaches can bring to bear on long-standing questions in GPCR and adrenoceptor biology. Although definitive answers to many questions are not yet possible, the increasing use of new approaches, from crystallography and structural biology to genetic epidemiology and population biology, seems destined to provide unexpected new insights and novel therapeutic agents that will stimulate, block, or modify the actions of adrenoceptors. Participants of the 2002 Satellite adjourned with eager anticipation of the discoveries that will be presented at a Seventh IUPHAR Satellite on Adrenoceptors, which we hope will be held in conjunction with the next IUPHAR meeting in Beijing, China (2006).
- © American Society for Pharmacology and Experimental Theraputics 2002

