REELIN and Schizophrenia:
A Disease at the Interface of the Genome and the Epigenome
- E. Costa, MD,
- Y. Chen, MD,
- J. Davis, MD,
- E. Dong, PhD,
- J.S. Noh, MD,
- L. Tremolizzo, MD,
- M. Veldic, MD,
- D.R. Grayson, PhD and
- A. Guidotti, MD
- Psychiatric Institute Department of Psychiatry, College of Medicine, University of Illinois at Chicago, Chicago, IL 60612
- EC. E-mail costa{at}psych.uic.edu; fax 312-413-4569.
Abstract
The downregulation of the Reelin gene (RELN) that occurs in schizophrenic brains, which are characterized by pyramidal neurons with shortened dendrites and by reduced expression densities of dendritic spines, may well result from hypermethylation of the RELN promoter. In the adult mammalian brain, γ-aminoburytic acid–secreting (GABAergic) interneurons release RELN into the extracellular matrix, where it binds with high affinity to the integrin receptors present at dendritic spine postsynaptic densities and likely plays a role, elaborated in this article, in synaptic plasticity. In heterozygous reeler mice, which are haploinsufficient in RELN, inhibitors of histone deacetylase increase DNA demethylase activity and restore RELN expression. Such inhibitors could thus be of therapeutic value in mitigating vulnerability to schizophrenia among high-risk individuals.

Introduction
The incidence of schizophrenia in the general population is about 1%, but is significantly higher in relatives of schizophrenics: the incidence is about 2% among third-degree relatives (e.g., first cousins and great grandchildren); 6% among second-degree relatives (e.g., nieces, nephews, and grandchildren); 17% in dizygotic twins; and peaks at about 50% in monozygotic twins (1). These statistics, moreover, confirm earlier adoption studies that reveal the vulnerability to schizophrenia to depend on the morbidity risk expressed by the biological parents to the exclusion of any risk associated with the adoptive parents (2).
A recent review by Lewis and Lieberman (3) not only supports the polygenic nature of schizophrenia, but also suggests that the morbidity risk begins to be expressed during embryonic development of the central nervous system (CNS). In particular, a number of abnormalities accompany early brain development of schizophrenic individuals, and the molecular neuropathology of schizophrenia is beginning to be unraveled. The autosomal recessive mouse mutant reeler has led to the identification of the protein Reelin (RELN) that is under investigation for its potential role(s) in the etiology of schizophrenia (Box 1).
Significantly, there is a great (~ 50%) deficit in the RELN content in specific areas of brains from schizophrenic individuals (4). Because this protein is normally operational during embryonic corticogenesis and throughout the patient's lifespan (see Box 1), the morbidity risk of schizophrenia, in accordance with the suggestion that embryonic factors contribute to the etiology of schizophrenia (3), may arise as a consequence of compromised RELN expression.
Reelin and Neurodevelopment
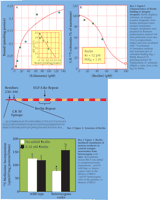
In the developing mammalian brain, the proper migration and localization of neurons depend on specific cell–cell interactions. For example, during corticogenesis, integrin receptors foster communication between primordial GABAergic neurons (i.e., Cajal-Retzius cells) and migrating pyramidal neurons that have not yet expressed synaptic links. Reelin (RELN) is a high-affinity ligand (picomolar range) of integrin receptors (Box 1, Figure 1⇓) and in the adult frontal cortex is synthesized by virtually every GABAergic neuron subtype (30, 46–48). Thus, we have argued that the primordial communication between GABAergic interneurons and pyramidal neurons that occurs via RELN agonistic action on integrin receptors (49) is in fact conserved throughout the mammalian lifespan. Specifically, in the cortex and hippocampus of the adult nonhuman primate, RELN is secreted by GABAergic neurons into the extracellular matrix and tends to bind to dendrites and dendritic spines of pyramidal neurons (48).
RELN is a 3461-residue protein that contains in its N-terminal domain the epitope of the CR50 antibody and also possesses eight EGF-like motifs (Box 1, Figure 2⇓) (42). The CR50 epitope binds to intergrin receptors expressed in dendritic spine postsynaptic densities (Box1, Figure 3⇓) and results in the intracellular phosphorylation of the Dab1 protein, an intracellular signal in the RELN pathway (50); this phosphorylation in fact is inhibited by the CR50 intibody (43). A second indication of RELN bioactivity is observed as an increase in rapamycin-sensitive protein synthesis when exogenous RELN is added to synaptoneurosomes prepared from brain homogenates of heterozygous reeler mice (Box1, Figure 3⇓). RELN-encoding mRNA includes a 5’ untranslated region that in primates includes a CGG repeat that is polymorphic, although no examined polymorphism (containing from seven to twenty-three repeats) correlates in humans to susceptibility to schizophrenia.
From an analysis of postmortem brains from four separate cohorts, it is apparent that the deficit of RELN that is associated with schizophrenia also extends to bipolar patients with mania, but not to unipolar patients without psychosis (5); appropriate controls indicate that neither the duration of the postmortem interval, the type of medication prescribed, nor a reduction in the number of cortical neurons determines the decrease of RELN expression in schizophrenia or bipolar disorder patients.
Anatomical studies of schizophrenia (6) and bipolar disorder patients (7) show that the density of dendritic spines in cortical pyramidal neurons is decreased, thereby suggesting that dendritic spine postsynaptic densities (DSPDs) and dendritic spine plasticity are compromised in both disorders. A similar compromise is observed in heterozygous reeler mice (8). A number of other anatomical and physiological similarities exist between the brains of heterozygous reeler mice and schizophrenics, including a reduction in both neuropil density and in sensory gating (8–11). Of course, studies of postmortem brains of schizophrenia patients cannot distinguish whether a deficit in the expression of a protein is a cause or a consequence of this disease.
THE HUMAN RELN GENE AND SCHIZOPHRENIA
A recent report convincingly shows that the null RELN mutation occurs in humans (12). The symptomatology of such individuals is dominated by lissencephaly (i.e., smooth brain) associated with a profound cognition deficit, a loss of muscle tone, and a radiologically evident brain deformation that is less severe but still abundantly expressed in heterozygous RELN persons. Thus, severe mental deficiency precludes a diagnosis of schizophrenia among humans bearing a RELN mutation. In addition, because the difference between the cognitive function of mice and humans is so profound, one cannot categorically assume the heterozygous reeler mouse to be an accurate model for schizophrenia. Furthermore, because the reeler mouse reflects a natural mutation, phenotypic traits cannot invariably be attributed to a deficiency of RELN. We are currently generating heterozygous reeler mice that express a RELN transgene to study whether the dendritic spine deficit can be reversed by expression of RELN. Nevertheless, the reduction of RELN expression that mitigates dendritic spine expression and dysfunction in heterozygous reeler mice remains relevant to molecular studies of schizophrenia, and to this extent such mice are valid tools in investigations of the disease.
REGULATION OF GENE EXPRESSION BY DNA METHYLTRANSFERASES
DNA methyltransferases (DNMTs) regulate gene expression by methylating cytosine nucleotides present in CpG islands of selected gene promoters. Resulting patterns of CpG methylation are recognized by MeCP2, a methyl-CpG–binding protein that likely mediates gene silencing and parental imprinting. With these mechanisms, several genes can be silenced in a cell-specific manner, and significant stretches of entire chromosomes can be silenced. These genomic patterns of gene silencing are maintained by DNMTs during cell replication (13).
In the vast majority of autosomal genes, paternal and maternal alleles are functionally equivalent. Nevertheless, scores of autosomal genes can be paternally or maternally imprinted, such that one of the parental alleles is silenced by hypermethylation of CpG islands so that the alternative parental allele alone is expressed (see 14). DNMT1 maintains parental imprinting via de novo methylation of imprinted genes in gametes (see 15). Several lines of evidence suggest that many imprinted genes are important for neuropsychiatric function (16).
Only recently has CpG methylation become appreciated as an important factor in the functionality and regulation of gene expression in the CNS. However, any appreciable degree of flexibility in the expression of CNS genes regulated in this manner would require the enzyme-catalyzed reversibility of cytosine methylation within particular gene sequences. This enzymatic requirement poses a conceptual problem in that the removal of methyl groups from cytosines is generally presumed to be energetically unfavorable, although base excision/nucleotide repair mechanisms may provide the means to a net demethylation reaction (17, 18), and the direct removal of the methyl group from methylated CpG dinucleotides, with concomitant methanol production, has also been demonstrated under certain circumstances (19). Further experiments are required to explain how the methyl-CpG binding domain of MeCP2, which is widely speculated to possess demethylase activity, may in fact contribute to the reversibility of DNA methylation in brain neurons. Indeed, DNA demethylating activity occurs in conjunction with histone hyperacetylation, brought about by the inhibition of histone deacetylase activity (see Box 2). It will be necessary to investigate such enzymatic partnerships in relation to RELN expression in primary cultures of GABAergic interneurons from neonatal mouse cortex that express RELN, DNMT1, and histone acetylase and deacetylase. We are now studying the GABAergic neuronal expression of DNA demethylating activity.
Histones: Functional Components of the Epigenome
The acetylation of specific lysine residues of histones H3 and H4 by histone acetyltransferase (HAT) causes a relaxation of chromatin supercoiling that can be reversed by the action of histone deacetylase. Histones and the components of the epigenome (defined as the layer of information that dictates which parts of the genome are expressed in a specific district and at specific time) act in a combinatorial fashion to activate or suppress the function of a given genomic region. Thus, the acetyl groups of histones that act to open chromatin arise from enzymatic functions that interact with the genome but are not part of it. The association of histones with DNA is reversible and can be modified pharmacologically (Figure 1⇓). The methylation of cytosine nucleotides within DNA represents another modification of the genome that regulates gene expression. In this case, the epigenome can be viewed as a coat of methyl groups; up to approximately 80% of CpG dinucleotides can be methylated. In general, regions of the genome that are transcriptionally active are less methylated than other regions. Intriguingly, methylated CpG sequences can be actively demethylated when they are packaged into hyperacetylated histones (see text; see 13).
As shown in Figure 1⇓, it is probable that deacetylase inhibitors that cause a persistent histone acetylation state create a condition that facilitates demethylation, as shown following the treatment with Trichostatin A and valproate. As components of the the epigenome, histones can thus function as targets for drugs that may modify the dynamic equilibrium of histone acetylation and deacetylation. Hydroxamic acid-based HDAC inhibitors have been developed that are effective in limiting tumor growth. It has been estimated that these agents act by altering transcription of less than 2% of genes expressed in tumor cells (51). Interestingly, various HDAC inhibitors show a certain degree of selectivity in terms of their mechanism of action (52). A key issue is the selectivity of various HDAC inhibitors on the expression of genes relevant to psychiatric disease.
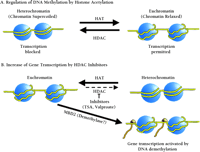
Proposed model for the transcriptional activation of genes regulated by methylation. Yellow line represents DNA and blue spheres indicate nucleosomes. Histone acetyltransferase (HAT); Histone Deacetylase (HDAC); Trichostatin (TSA); Methyl-CpG-binding Domain 2 (MBD2).
METHIONINE-MEDIATED EXACERBATION OF SCHIZOPHRENIA
In efforts to assign a role to dopamine in the etiology of schizophrenia, many studies examined the effects of high doses of methionine, either alone or along with monoamineoxidase inhibitors, on the disease (20–29). By providing the brain with methionine, a precursor of the methyl donor S-adenosylmethionine (SAM), the hypothesis to be tested was whether dopamine inactivation (i.e., the methylation of dopamine via catechol-O-methyltransferase (COMT)) could mitigate the disease. As an extra dividend, it was hoped that this treatment could help explain the beneficial effects of chlorpromazine and haloperidol, two dopamine receptor antagonists.
In Table 1⇓, we summarize the results reported by Antun et al. (22) following the administration of high doses of methionine. Clearly, these data indicate that methionine in fact exacerbates schizophrenia symptomatology in about 67% of patients, but does not cause a “chronic brain syndrome” as suggested by Cohen et al. (29). Furthermore, the report of Antun et al. (22) argues against an increase in dopamine metabolites via COMT activation and should, moreover, alleviate any concern that methionine causes neurotoxicity.
Because increased levels of methionine—and presumably the concomitant increased levels of the methyl donor SAM—failed to increase dopamine turnover by COMT, we decided to test whether the exacerbation of symptomatology was driven by increased DNMT1 activity, which, we hypothesized, would increase the methylation of CpG island-containing promoters, such as the RELN promoter.
DNA METHYLTRANSFERASE AND RELN LEVELS IN THE FRONTAL CORTEX: TOWARDS A MOLECULAR DEFINITION OF SCHIZOPHRENIA
In neural progenitor NT2 cells, addition of the DNA methyltransferase inhibitor 5-azadeoxycytidine (AzadC) can increase the expression of RELN by 20-fold (Table 2⇓) or more, suggesting that the RELN promoter is normally (i.e., in the absence of AzadC) hypermethylated. To explore this possibility, we measured the levels of mRNA for DNMT1 by in situ hybridization of various layers of the frontal cortex from nonpsychiatric and schizophrenic patients (Table 3⇓). The number of neurons of DNMT1-encoding mRNA, in several cortical layers, was increased in schizophrenia patients relative to nonpsychiatric subjects. Significantly, in most of the cortical layers, the increase in neurons producing DNMT1-encoding mRNA coincided with a decrease in the number of RELN-positive neurons.
RELN- and DNMT1-encoding mRNAs are not expressed in pyramidal neurons, but are expressed in small cells that can be considered interneurons because they react with an antibody for a neuronal nuclear protein. Although it is impossible to morphologically differentiate GABAergic neurons in layer IV, there is a great probability in the other cortical layers that virtually all non-pyramidal neurons are GABAergic. Thus, because there is no significant difference, in any cortical layer of control subjects, in the number of neurons expressing the two types of mRNA, we can suggest that DNMT1 and RELN are selectively expressed in GABAergic interneurons in the frontal cortex; however, we have only proven that RELN is consistently coexpressed with a label specific for GABAergic neurons (30). It is very important to establish whether the same GABAergic neurons express both RELN and DNMT1 because such coexpression would offer a unique opportunity to study genomic methylation and epigenomic histone hyperacetylation and deacetylation within given, RELN-secreting GABAergic neurons.
In cortical layers I, II, IV, and V of schizophrenia patients, the number of neurons positive for RELN-encoding mRNA is reduced relative to nonpsychiatric subjects, whereas the number of neurons expressing the DNMT1-encoding mRNA is lower in the nonpsychiatric subjects (Table 3⇓). These results suggest that a higher number of neurons positive for DNMT1, along with fewer neurons positive for RELN, will be an essential characteristic of schizophrenia neuropathology. Moreover, these results are consistent with the possibility that hypermethylation of the RELN promoter may be pathogenic. Histone hyperacetylation due to the inhibition of histone deacetylase (31) may also regulate the demethylation of cytosines in CpG islands (32). The recent recognition of such pharmacolgically induced epigenomic–genomic interactions has offered new possibilities for a pharmacological modification of epigenetic hypermethylation of promoter function (see Box 2).
THE EPIGENETICS OF REDUCED RELN LEVELS IN SCHIZOPHRENIA
Heterozygous reeler mice have been regarded as a mechanistic model to study RELN downregulation in schizophrenia (8). However, as stated above, the validity of such a model cannot be confirmed in the few heterozygous RELN humans that have been studied (12), where the concomitance of mental deficiency and brain structural abnormalities prevent a clear diagnosis of cognitive disorder. So, does a suitable model exist for epigenetically induced downregulation (via hypermethylation) of RELN in schizophrenia?
Table 4⇓ shows that the injection of methionine for fifteen days into wild-type and heterozygous reeler mice decreases the expression of RELN in the frontal cortex. Thus, this observation suggests that environmental factors that increase SAM content may also downregulate RELN expression. As seen in Table 3⇓, expression of DNMT1in human brains appears to be highly compartmentalized, but we do not know whether SAM or the mRNA that encodes methionine adenosyl transferase 2 (the enzyme responsible for SAM synthesis in brain) is also present in GABAergic neurons. The answer to this question will further help us to establish whether a defect of GABAergic neuron function contributes to the etiology of schizophrenia.
INHIBITION OF HISTONE DEACETYLASE
Box 2 suggests a pharmacological intervention to sustain RELN expression as a hypothetical means of mitigating schizophrenia (33–34). Mice injected for fifteen days with valproic acid, a weak inhibitor of histone deacetylase and a drug used as a mood stabilizer in psychiatry, show an increase in levels of RELN-encoding mRNA (Figure 1A⇓). Curiously, levels of translation product (i.e., 400-kDa RELN) fail to increase, although levels of 50-kDa RELN metabolites are increased (Figure 1B⇓). This apparent instability of RELN may be due to the deficit of binding sites for RELN (i.e., a deficit of integrins or other proteins associated with dendrites and dendritic spines) demonstrated in heterozygous reeler mice (8–9). Moreover, as seen in Table 2⇓, Trichostatin A, a potent inhibitor of histone deacetylase, significantly increases the accumulation of RELN-encoding mRNA in NT2 cell cultures.
GENOMIC AND EPIGENOMIC FACTORS IN NEURONAL PLASTICITY
DNA methylation is a modification of the genome that is emerging as a major mechanism by which the environment (e.g., hormones and extracellular matrix signals such as RELN) epigenetically affects transcriptional regulation. In this context, the methylation status of DNA can be influenced so as to affect gene expression programs that establish long-lasting changes in specific neuronal circuits associated with learning and memory. It is thus crucial to understand 1) how DNA methylation can change gene expression, and 2) how DNA methylation patterns are changed or maintained via DNA demethylase inhibition or activation. The latter question requires a more detailed understanding of demethylase activation. The definition of the genomic and epigenetic interactions in plasticity must consider the role of RELN in DSPD-regulated protein synthesis (Box 1, Figure 3⇑) and in the remodeling of brain structure and function related to learning and memory.
In studying defects of cortical function such as schizophrenia and bipolar disorders, the brain's ability to acquire structural and functional individuality via dendritic and DSPD plasticity takes center stage. Neuronal plasticity was initially proposed to help understand the mechanisms of CNS repair following neuronal damage; however, DSPD plasticity is now considered the very key to individuality in cognitive function. Changes in brain structure are caused by environmentally introduced plastic modifications that can be short (seconds or minutes) or long lasting (hours, days, or years). Long-lasting plastic responses require that new proteins be synthesized during an initial phase that assures the precise definition of the plastic response. Due to the length of axons and the complexity of cortical and hippocampal pyramidal neuron dendritic arborization, protein synthesis is extrasomal, occurring at the sites where nascent proteins will function in dendrites or axon terminals (35–36). It is now believed that the translation of mRNAs resident in dendritic spines (37) is activated by a complex signaling mechanism leading to the phosphorylation of a cytoplasmic polyadenylation binding element (CPEB) that activates a polyadenylate polymerase (38). In this mechanism, the mammalian target of rapamycin (mTOR) (39) and RELN play an important role (see Box 1). In fact, the integrin receptors that mediate the action of RELN and mTOR are essential to these extrasomal translational events. One should emphasize that the head of dendritic spines, where postsynaptic densities are located, function as a convergence site of all excitatory afferent information. DSPDs are therefore signal transduction centers that modify dendritic spine volume, shape, and function, and are capable of expressing new protein synthesis (40). They pilot the expression of brain circuit diversity, contributing to intellectual individuality.
CONCLUSIONS
The prevailing view is that schizophrenia is a “two-hit” neurodevelopmental disorder in which structural brain changes caused by a prenatal or perinatal insult (i.e., the first “hit”) confer a predisposition to the disease. In fact, to the extent that brain development is a life-long plastic process (41), with structural modifications occurring during long-lasting responses to environmental stimuli associated with learning, it follows that schizophrenia encompasses molecular processes initiating prenatally and operative throughout the life of the patients.
Thus, schizophrenia cannot reflect discrete insults to merely one or several genes, because we know that the illness is a life-long disabling condition for only about 40% of patients. Hence, the pathophysiological mechanisms involved in schizophrenia might include defects in a slew of promoters regulated epigenetically by CpG methylation. RELN fits readily into this paradigm; its expression is controlled by the interaction between genome and the epigenetic “histone code.” We believe that RELN happens to be deficient in schizophrenia (5) because of an epigenetic hypermethylation of its promoter region. Others have furnished evidence of the important neurodevelopmental role of RELN in corticogenesis and perhaps in thalamus, neostriatum, and spinal cord function (42–45). We have shown that RELN is downregulated in schizophrenia (5), and have posited an important physiological role for RELN in regulating extrasomal protein synthesis important for dendritic spine plasticity. Of course, epigenetic methylation in schizophrenia may apply to many other genes as well, such as those involved in transmitter biosynthesis or postsynaptic receptor biogenesis.
It is time to begin to think in terms of pharmacological interventions that include inhibitors of histone deacetylase and of other modifiers of the epigenetic DNA methylation mechanisms that appear to be operative in the RELN deficit of schizophrenia. The role of RELN in dendritic spine plasticity would be in line with the disordered transcortical connectivity that is now considered to be a core problem in schizophrenia.
l-Methionine–Induced Aggravation of Schizophreniaa
Increase in RELN mRNA after Treatment with Inhibitors of Histone Deacetyase
Comparisons Between RELN-and DNMT1-Positive Neuronal Counts in Prefrontal Cortex (Brodmann area 10)
Protracted Treatment with Methionine (1g/kg twice daily for 15 Days) Decreases Reelin Protein and mRNA Expression in Frontal Cortex of Wild-Type Mice (wtm) And Heterozygousreeler Mice (hrm).
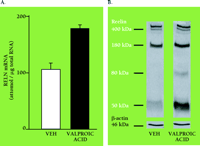
RELN mRNA and RELN metabolites (50 kDa) in frontal cortex are increased following treatment of heterozygous reeler mice with valproic acid. Valproic acid (100 mg/kg) was administered subcutaneously twice a day for 15 days. A) RELN mRNA was measured by quantitative RT-PCR (4); each value is the mean ± SEM of 5 animals; B) RELN-like immunoreactive peptides; representative western blot analysis of vehicle (VEH) and valproic acid treated animals.
Acknowledgments
This work was supported by grants from the National Institutes of Health MH062090 to EC and MH062188 to AG.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

E Costa, MD
Y. Chen, MD

J.M. Davis, MD

E. Dong, PhD

J.S. Noh, MD

L. Tremolizzo, MD
M. Veldic, MD

D.R. Grayson, PhD

A. Guidotti, MD

