Coming of Age:Anti–Cytokine Therapies
- Xiao-yu R. Song, PhD,
- Theodore J. Torphy, PhD,
- Don E. Griswold, PhD and
- David Shealy, PhD
- XyS. E-mail songx{at}centocor.com; Fax 610-651-7363.
Abstract
Although cytokines are critical in maintaining normal physiology, excessive production of these proteins can lead to pathological consequences. Inhibitors of cytokine production or action are therefore widely investigated as potential therapeutic agents in a variety of immune and inflammatory diseases. Indeed, the successful application of inhibitors of tumor necrosis factor-α in rheumatoid arthritis and Crohn's disease heralds the great therapeutic potential of biopharmaceutical agents to counteract cytokine activities. Emerging opportunities for new therapeutics, as well as the challenges we face in their use and development, are described in this review.

Cytokines are polypeptide regulators of intercellular communication within the immune system. The cells depicted above are
macrophages and T-lymphocytes responding to cytokines (green rods). The Y-shaped structures represent antibody-derived therapeutics
that recognize cytokines, either in circulation or bound to cell surface receptors, to block their action. Therapeutic strategies
that interfere with cytokine function may be effective in immune disorders such as asthma, cancer, arthritis, and transplant
rejection.
INTRODUCTION
Biopharmaceuticals have an important role in the treatment of a variety of disorders. In particular, several “biologics” are strikingly effective in treating specific cardiovascular diseases, autoimmune disorders, and cancers that are resistant to conventional pharmacotherapy. The success of biopharmaceuticals is the result of decades of promising starts followed by demoralizing failures. But now, these agents are an accepted part of medical practice. Indeed, for the aforementioned diseases, their use is nearly commonplace and likely to expand. Illustrative of this point is the fact that ten monoclonal antibodies are currently approved for marketing, with more than seventy additional agents in clinical development (1).
Historically, small-molecule approaches have not been successful in disrupting cytokine action. Simplistically, the explanation for this failure is twofold. First, protein–protein interactions, such as those that occur with the binding of a cytokine to its receptor, are generally difficult to disrupt with small molecules. Second, small-molecule drugs that target downstream cytokine signal transduction pathways are notoriously nonselective and, consequently, evoke a broad range of toxicities. In contrast, biopharmaceuticals can readily disrupt interactions between cytokines and their receptors, and can do so in an exquisitely selective manner. This review focuses on the use of cytokine-inhibiting biologics recently approved as therapeutics, and on some that are now emerging from clinical development.
STRATEGIES TO INHIBIT CYTOKINE ACTIVITY
Antibodies
A leading approach to neutralize cytokines is the use of specific antibodies directed against the cytokine or its corresponding receptor. Antibodies have several central features that are important in strategies to interfere with cytokine action. These include excellent solubility, exquisite specificity, and a long half-life in serum.
Kohler and Milstein's landmark method (2) for the generation and selection of hybridoma cell lines to produce specific, monoclonal antibodies has been revolutionary. This procedure allows the unlimited production of specific antibodies that can neutralize self-antigens such as cytokines. Specifically, hybridomas are produced by fusing murine myeloma cells with splenocytes, thereby generating immortal cell lines, each of which produces a specific “monoclonal” antibody (i.e., a unique protein molecule; Figure 1⇓). By screening sufficient numbers of clones, it is possible to select an immortal cell line that produces antibody with the desired antigen specificity and Fc functionality. The murine anti-interleukin(IL)-6 monoclonal antibody BE-8 (Figure 2A⇓), used to treat B-lymphoproliferative disease and multiple myeloma, is limited clinically to treatment regimens of no greater than two weeks, due to the classic immune response elicited in patients against murine proteins (3). Therapeutic limitations arising from the immunogenicity of foreign antibodies, however, can be mitigated by recombinant DNA technology. A major advance in this respect was the generation of human–murine chimeric antibodies, in which the constant regions of the antibody are of human origin and the antigen-binding function of the chimera is of murine origin (4). An example of a clinically useful chimeric antibody is REMICADE® (Figure 2B⇓), which attains its antigen binding properties by virtue of a murine variable sequence but contains a human-derived constant region, so that the immunogenicity of the chimeric molecule, when administered to humans, is significantly mitigated relative to the corresponding murine antibody (cf. Figures 2A versus 2B⇓).

Generation of monoclonal antibodies. B-lymphocytes from immunized mice (or transgenic, “humanized” mice) are fused with myeloma cells. Subsequently, the cell producing the antibody of interest is selected. Antibodies are characterized by two Fab domains which contain the complimentarity-determining regions (CDR) that provide antigen specificity, and an Fc domain which contains the binding sites necessary for complement fixation and other functions.
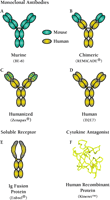
Categories of biologics that can target cytokines. Cytokine-blocking biologics include antibodies and receptor-derived constructs, and natural antagonist proteins. Clinically successful examples from each category are given: A-D, monoclonal antibodies; E, an immunoadhesin (consisting of two extracellular cytokine receptor domains fused with immunoglobulin constant heavy domains); and F, recombinant form of the naturally occurring IL-1 receptor antagonist. See text and Table 1 for details.
Antibody generation can be further refined for clinical use by replacing the murine variable domain framework with corresponding human sequences, so that the complementarity-determining regions (CDRs) are the sole murine sequences contributed to the antibody molecule (5). Although replacing the murine variable components reduces immunogenicity, these “humanized” antibodies (Figure 2C⇑) tend to have a lower affinity for antigen than the analogous murine antibody, so that site-specific mutations in the CDRs themselves are sometimes necessary in order to optimize affinity for antigen. Clinical studies with humanized antibodies such as daclizimab (i.e., Zenapax®; Figure 2C⇑) show, as do chimeric antibodies, a clear improvement over murine antibodies in terms of both immunogenicity and half-life in serum (6).
Two recent developments allow the isolation of fully human antibodies directed against virtually any antigen. The first of these exploits transgenic mice (7). The XenoMouse® technology, developed by Abgenix, and the HuMAb-Mouse® technology, offered by Medarex, introduce human immunoglobulin gene sequences into mice that are compromised with respect to murine antibody generation. Such mice can be immunized with an antigen of interest, and the immune systems of these laboratory mice respond by generating fully human antibodies. Hybridoma cell lines can then be produced from the mouse splenocytes and screened for production of human antibody with the desired characteristics (see Figure 1⇑).
Phage display is a second development that has advanced clinical interests in human monoclonal antibodies. In this technology, experimental genes of interest are fused with a phage gene that encodes a coat protein. Such gene constructs, when placed within the bacteriophage genome, produce fusion proteins that are “displayed” on the surface of the phage particle. In the early 1990s, Griffiths and colleagues (8) showed that repertoires of human antibody variable domains (both heavy and light chains), expressed from cloned genes derived from a large population of human B-cells, are amenable to phage display. Specifically, high numbers of variable domains were constructed such that each phage displayed a distinct, single-polypeptide “variable domain” arising from the fusion of one variable heavy and one variable light chain sequence. An extensive library of phage could thus be screened for antigen binding and then replicated upon inoculation into bacteria (Figure 3⇓).
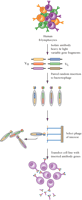
Generation of a phage display library for antibody selection and production. Human B lymphocytes are the source of a repertoire of antibody heavy and light chain gene segments (VH and VL) that are inserted in a pair-wise, random manner into a bacteriophage genome. Mature phage expresses the VH–VL polypeptides fused to phage coat proteins. Phages that express the desired VH–VL polypeptide can be selected and the corresponding antibody genes inserted into a cell line for expression and production of antibody.
The great advantage of phage display is that it eliminates the need to immunize an animal; however, the array of antigen-binding proteins amenable to phage display is limited by the number of sequences compiled within the original library of variable gene constructs. To circumvent this limitation, a completely synthetic version of an immunoglobulin phage library has been developed with the aim of expanding the diversity of available variable gene combinations (9).
Immunoadhesins
Another approach used to develop biologic inhibitors of cytokine activity is to engineer a fusion protein that combines the constant domains of an antibody molecule with the ligand-recognition domain of a cytokine receptor (Figure 2E⇑). Such fusion proteins, or immunoadhesins, can be expressed and secreted from mammalian cells as bivalent proteins that, much the same as immunoglobulins, consist of two polypeptide chains that are disulfide linked (10). The advantages of this approach is that it not only eliminates the need to immunize an animal, but also circumvents screening for cytokine-specific antibodies: “antigen recognition” is provided by the cytokine receptor that is engineered into the immunoadhesin. Immunoadhesins in general share the extended half-life in serum observed for whole antibodies, and because they are derived from human proteins, they tend to manifest little immunogenicity in the clinic.
Although many immunoadhesins have been prepared for research studies, only a few have entered clinical trials, and only one is approved for human use (i.e., Enbrel®). Enbrel® (etanercept) consists of two extracellular domains of the p75 form (i.e., type II) of the TNF receptor fused with the constant domains of an IgG1 heavy chain. The half-life of etanercept in serum is reported to be 4.8 days, and an immune response is observed in about 16% of treated patients, according to the Enbrel package insert. An immune response presents a unique challenge for immunoadhesins, even if the antibodies are non-neutralizing. Because TNF-α and other cytokines activate specific signaling pathways after binding to cell membrane–associated receptors, antibodies directed against the receptor moiety may be able to bind native cell receptors and thereby activate signaling pathways (11). Therefore, patients who develop an immune response against immunoadhesins could potentially produce antibodies with agonist activity.
Natural Cytokine Antagonists
Cytokines expressed during an inflammatory response are tightly controlled at the transcriptional and translational levels, and they appear to act locally in that they are cleared rapidly once they diffuse into the vascular compartment. Mechanisms have also evolved to allow rapid neutralization of excess cytokine at the site of inflammation, such as the expression of soluble cytokine-binding proteins found in tissue and blood. Other mechanisms for regulating cytokine-induced pathways include natural antagonists. In the case of interleukin (IL)-1, a soluble form of IL-1 has been described that can bind with high affinity to the cell surface receptor without activating signaling (12). This protein, called IL-1 receptor antagonist, and marketed as a recombinant biologic called Kineret® (anakinra; Figure 2F⇑), competes with IL-1 for binding to receptor.
THE ROLE OF CYTOKINES IN THE PATHOGENESIS OF IMMUNE AND INFLAMMATORY DISEASES
As a family of bioactive proteins and polypeptides synthesized by white blood cells and virtually all other nucleated cells (13), cytokines are secreted in response to microbes and other antigens, as well as environmental stimuli, and thus mediate diverse biological processes required for homeostasis and host defense. These processes include immune responses, inflammation, cell growth, tissue repair, fibrosis, and angiogenesis. Cytokines play critical roles in host defense against pathogens and provide links between innate and adaptive immunity. They also regulate the magnitude and the nature of immune responses by influencing the growth and differentiation of immune cells.
TNF-α and IL-1 in Rheumatoid Arthritis and Crohn's Disease
TNF-α and IL-1 are present in the rheumatoid joint fluids and synovial membranes of patients with rheumatoid arthritis (RA), and play a crucial role in the pathogenesis of the disease (14), by mediating leukocyte recruitment and synovial inflammation. Both recruited and resident synovial cells subsequently release a host of proinflammatory mediators, which leads to the activation of osteoclasts and induction of bone and cartilage breakdown. Both TNF-α and IL-1 directly activate osteoclasts, or indirectly enhance osteoclast activity through the induction of IL-6, IL-11, IL-15, IL-17, and a host of proteolytic enzymes. Similarly, TNF-α and IL-1 also cause cartilage degradation, either directly by stimulating the synthesis of collagenases in articular chondrocytes, or indirectly by activating synovial fibroblasts, which subsequently secrete various enzymes including matrix metalloproteases. That TNF-α is a central mediator in numerous pathophysiological processes is known from studies showing that anti-TNF-α antibody inhibits the generation of IL-1, IL-6, IL-8, and granulocyte macrophage colony stimulating factor in rheumatoid synovial cultures (15). Studies in animals further support the role of TNF-α in RA. In particular, administration of an anti-TNF-α antibody ameliorates collagen-induced arthritis (16). Collectively, these studies provide powerful evidence that TNF-α is a primary mediator of RA as well as other immune and inflammatory diseases (Figure 4⇓).
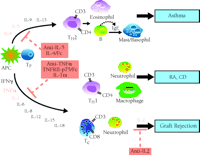
A panoply of cytokines exist that can be targeted in disease states such as asthma, rheumatoid arthritis (RA), Crohn's disease (CD), and graft rejection. Major cell types involved in disease pathogenesis are also depicted. See text and Table 1 for details.
Similarly, several lines of evidence suggest that TNF-α plays a central role in the pathogenesis of mucosal inflammation in Crohn's disease (CD), and is likely to be at the apex of the inflammatory cascade. In CD, an excessive activation of macrophages and T helper 1 (Th1) cells leads to the enhanced production of proinflammatory cytokines such as TNF-α, IL-1, IL-6, IL-8, IL-12, and interferon-γ in inflamed intestinal mucosa (Figure 4⇑) (17); increased expression of TNF-α in particular, both at the level of translation and transcription, characterizes mucosal biopsies from patients with CD. Thus, the cytokine milieu in CD favors an amplification of the inflammatory cascade and secretion of more inflammatory mediators, destructive enzymes, and free radicals that cause tissue injury, mucosal permeability, diarrhea, and fibrosis (18). Both human and animal studies indicate that TNF-α plays a crucial role in establishing pathogenesis. For example, the overexpression of TNF-α in mice results in the development of a “Crohn's-like” phenotype, and anti-TNF-α treatment ameliorates intestinal inflammation in several animal models. Finally, and quite significantly, CD patients respond to treatment with a single infusion of the monoclonal anti-TNF-α antibody marketed as REMICADE® (19).
Cytokines in Asthma
Asthma is a pulmonary disease associated with an allergic inflammatory response to a variety of antigens. The disease is characterized by increased pulmonary inflammation followed by bronchial hyperreactivity and, eventually, decreased lung function. Among a host of mediators linked with the pathophysiology of asthma, the cytokines produced by T 2 helper cells (Th2) are considered central players; these include IL-4, IL-5, IL-9, and IL-13 (Figure 4⇑) (20). IL-4 facilitates the adhesion and transmigration of eosinophils into the lungs through the upregulation of adhesion molecules on the pulmonary vascular endothelium. Moreover, IL-4 stimulates mast cell growth and also plays a role in inducing mucus production from pulmonary epithelium. In addition, IL-4 is a major regulator of IgE production by B cells, and is required for optimal Th2 differentiation.
Although IL-4 and IL-13 produce many of the same biological effects, IL-13 appears to be the more potent mediator of experimental models of asthma. Mice exposed to IL-13 on a chronic basis have all the hallmark features of acute challenge, such as airway hyperresponsiveness, increased mucus production, smooth muscle hyperplasia, IgE production, eotaxin production, and eosinophil infiltration. In the airways, moreover, these mice manifest fibrosis and the intense airway deposition of a crystalline substance, both of which are commonly noted in asthmatic patients. In asthmatic patients, IL-13 levels are increased in bronchial lavage fluid, which also correlates with eosinophil infiltration (21). Therefore, IL-13 seems to play an important role in the effector phase of the asthmatic response. In contrast, IL-5 acts mainly on eosinophils, stimulating differentiation and migration to the lung. IL-5 also activates eosinophils to produce cytotoxic agents, and can cause bronchial epithelium damage that may lead to hyperreactivity of the underlying bronchial smooth muscle.
IL-9 is another modulator in the pathogenesis of asthma. The in vivo evidence for the role of IL-9 was first determined in animal models of airway inflammation, where a close association was detected between the expression of IL-9 and bronchial hyperresponsiveness (22). Additionally, IL-9 protein levels in the lung correlate well with levels of airway responsiveness in mice. Moreover, selective overexpression of the IL-9 gene within the lungs of transgenic mice result in massive airway inflammation, along with infiltration of eosinophils and lymphocytes, mast cell hyperplasia, and increased subepithelial collagen deposition (23). More importantly, accumulation of mucus-like material within nonciliated cells accompanies goblet cell hyperplasia and a marked increase in airway responsiveness to inhaled methacholine. More recently, examination of bronchial biopsies show a highly significant increase in the expression of IL-9 in the airways of asthmatic subjects as compared to healthy control subjects (24). Not surprisingly, many new therapies in development for the treatment of asthma are aimed at inhibiting components of the allergic inflammatory response.
IL-2 in Organ Transplantation
IL-2 is a cytokine produced by T cells to promote the growth and differentiation of T and B cells. IL-2 also plays a crucial role in the inflammatory events that culminate, in the absence of immunosuppressive drugs, in the rejection and destruction of tissue transplants. For instance, in experimental models, the induction of IL-2 precedes acute graft rejection, and in humans, IL-2–encoding mRNA accumulates prior to acute rejection (25). T cells release IL-2 in response to allograft antigens that are presented to them by specific, antigen-presenting cells of the host immune system. IL-2 subsequently stimulates the expansion of clonal T cells that are specific for the allograft antigens, and these cells, in turn, release more IL-2, resulting in an amplified inflammatory response (Figure 4⇑); the amplification process also acts to mobilize other T cells to attack the allograft. Thus, clinical interventions that target IL-2 represent a more selective therapeutic strategy than general immunosuppression, because only committed and activated T cells are thereby targeted.
ANTI-CYTOKINE THERAPIES: CLINICAL EXPERIENCE
Rheumatoid Arthritis and Crohn's Disease
A number of biopharmaceuticals have been produced to interfere with cytokine activity (Table 1⇓), and a few have entered into clinical practice for the treatment of immune-mediated inflammatory diseases. Two pioneer biopharmaceuticals are infliximab (REMICADE®; Centocor), approved for treatment of CD and RA, and etanercept (Enbrel®; Immunex), approved for treatment of RA and psoriatic arthritis. Because these agents may lay the trail for future breakthrough biologic therapeutics, their profiles are discussed in detail.
Development of anti-cytokine therapeutics
Etanercept
As described above, etanercept, an immunoadhesin produced in mammalian cells by recombinant DNA technology (Figure 2E⇑), inhibits TNF-α activity by competing against cell surface TNF receptors for TNF-α. In addition, soluble TNF receptors can bind to lymphotoxin (LT)-α (also known as TNF-β). The efficacy of etanercept in RA has been demonstrated in several controlled clinical trials (26, 27). Fifty-nine percent of the patients receiving etanercept (25 mg) twice weekly achieved a 20% American College of Rheumatology (ACR) response, compared to 11% in the placebo group, after six months of treatment (27). A recent one-year study showed that etanercept (25 mg) given twice weekly is as efficacious as methotrexate in controlling early signs and symptoms of RA (28). When progression of joint damage was measured radiologically, no significant difference was noted between the two treatment groups; however, new joint erosions were significantly lower in the patients receiving etanercept as compared to those receiving methotrexate. In addition, no difference was detected in joint space narrowing.
Current treatment for juvenile RA (JRA) relies on many of the same drugs used on adults. Although methotrexate can benefit patients with JRA, many do not respond adequately, and there is concern about the side-effects in young patients. A clinical study of severe polyarticular JRA patients shows that 0.4 mg/kg etanercept administered twice weekly for ninety days results in improvement for 74% of patients (29). After an additional nine months of treatment, half of the patients who were randomized to receive etanercept had a disease relapse rate of 24%, in contrast to a rate of 77% for those receiving placebo. Etanercept has subsequently been approved for the treatment of JRA. In addition, etanercept has also been approved for treatment of psoriatic arthritis. According to a twelve-week randomized, double-blind, placebo-controlled study, 73% of etanercept-treated patients may achieve a 20% ACR response, compared with a 13% response for placebo-treated patients. Moreover, 26% of psoriasis patients have can achieve 75% improvement, in terms of psoriasis area severity index, whereas no improvement is observed for placebo-treated patients (30). Thus, etanercept offers an alternative to methotrexate in the control of JRA and psoriasis. In contrast, subcutaneous etanercept (25 mg twice weekly) is not effective in treating patients with moderate to severe CD (31).
Infliximab
The first clinical studies of infliximab in treating RA were carried out at the Kennedy Institute of Rheumatology in London, England, as an open, uncontrolled trial in which a single infusion of infliximab (20 mg/kg) was given to long-standing active RA patients who had failed all prior therapies. The single infusion of infliximab alleviated symptoms and reduced the numbers of swollen and tender joints for two to four weeks (14). Perhaps most significantly, a randomized, placebo-controlled phase III study, designated ATTRACT (anti-TNF therapy of rheumatoid arthritis with concomitant therapy) (32), confirmed that repeated infusions of infliximab affords incremental improvement in patients with active RA despite methotrexate therapy. Infliximab-treated patients, relative to the methotrexate control group, showed generally rapid and significant improvement by two weeks, and the great majority achieved a 20% ACR response within six weeks. Moreover, the number of responders increased over time. The infliximab-treated groups ultimately achieved 60-70% improvement in disease activity as compared to the placebo group. More importantly, there was strong evidence from radiographic assessment of hands and feet (in week 54 of treatment) that infliximab prevented progression of joint damage, including joint space narrowing and bone erosion. More interestingly, this structural benefit was observed not only in patients who had achieved the 20% ACR response, but also in patients who failed to do so. Finally, the joint-protective effects of infliximab in combination with methotrexate was maintained through two years of analysis.
For the treatment of CD, infliximab has become, on the basis of three landmark clinical trials, the sole anti-TNF-α agent to receive regulatory approval. The first trial confirmed early indications of symptomatic relief and mucosal healing, upon treatment with infliximab, in patients with refractory disease (33). Response rates (i.e., decreases of 70 points or more on the CD activity index) in patients who received infliximab (5 mg/kg) reached 81%, compared to 17% in the placebo group; indeed, over 50% of treated patients achieved clinical remission, compared to 4% of control subjects. This study unequivocally demonstrates the short-term efficacy of a single infusion of infliximab in treating active CD. In a follow-up trial, multiple infusions of infliximab allowed patients to maintain their initial response, whereas the placebo group gradually lost the response. Similar results were reported for other end points, such as clinical remission, quality-of-life measurement, and levels of C-reactive protein (34).
A third, pivotal trial reveals that infliximab can more than double the rates at which patients experience significant reduction (i.e., by half or more) in the number of draining fistulae associated with CD (35). Complete closure of the fistulae was observed in 55% of the patients receiving infliximab (5 mg/kg), compared to only 13% of patients in the placebo group. These results conclusively demonstrate the ability of infliximab to heal enterocutaneous fistulae in CD.
TNF-α Antagonists
Studies in TNF-α knockout mice and other animal models indicate that the very role of anti-TNFα agents in preventing immune activation (Figure 4⇑) also increases the risk of infection, including the reactivation of latent tuberculosis (36). However, appropriate evaluation of benefit-to-risk ratios, and screening measures as directed on the infliximab product label, should allow good management of infection risks. Similarly, because rare cases of demyelinating disorders have been described in etanercept-treated patients, special vigilance is called for with regard to preexisting or recent-onset central nervous system disorders.
Anakinra
Anakinra (Kineret®; Amgen) has been evaluated in several clinical studies of RA and reduces both the clinical manifestations of arthritis and the rate of progressive joint damage. In the first randomized, double-blind, placebo-controlled study, 472 RA patients receiving daily subcutaneous injections of anakinra (150 mg) for twenty-four weeks (37). manifested, relative to the placebo group, approximately 35% improvement in various clinical parameters; in particular, the frequency and severity of radiological joint erosions was reduced. Moreover, when anakinra was appropriately used in combination with methotrexate, the rate of individuals achieving a 20% ACR response (42%) was nearly doubled relative to the rate in patients receiving only methotrexate and placebo (23%) (38). Five percent of patients had to be withdrawn from treatment, due to injection site reaction, and an increased incidence of serious infections (2%) relative to the control population (1%) was also observed; anakrina is therefore contraindicated for patients with active or serious infections.
Asthma
Increasing knowledge of the pathophysiological roles of various cytokines in asthma has prompted the evaluation of novel anti-cytokine therapies. The proinflammatory cytokines IL-4, IL-5, and IL-13 are among the therapeutic targets (Table 1⇑). A soluble recombinant human IL-4 receptor has produced clinical benefits for patients with moderate asthma who daily require inhaled corticosteroids (39). Results with a humanized anti-IL-5 antibody, however, were disappointing. Although the antibody markedly reduces the number of circulating eosinophils and prevents their accumulation in airways, it is unable to affect early and late allergic responses, and fails to reduce airway reactivity to methacoline (40). Agents that target IL-13 are still early in the development process and thus remain to be evaluated in asthma.
Organ Transplant
Daclizumab
Daclizumab (Zenapax®; Protein Design Labs/Hoffman LaRoche) is one of two new prophylactic therapies for renal allograft rejection (Table 1⇑). It is a humanized monoclonal antibody consisting of murine CDRs that recognize IL-2R (CD25) and are fused to human IgG1 constant and variable frame-work domains. The efficacy of daclizumab has been demonstrated by two phase-III studies (41, 42). Daclizumab reduces the frequency of biopsy-documented acute rejection during the first six months after transplantation by approximately 40%. In addition, daclizumab-treated patients have fewer episodes of rejection, and the time to rejection is significantly delayed. Daclizumab has not been associated with any immediate side effects. Some evidence (43) indicates, moreover, a significant improvement in one-year survival rates for daclizumab-treated patients.
Basiliximab
Basiliximab (Simulect®; Novartis) is a chimeric monoclonal antibody consisting of the human IgG1κ Fc region fused with the Fab domain of a murine anti-human IL-2R antibody (Table 1⇑). Basiliximab has been evaluated in two phase-III clinical trials (43, 44). In general, basiliximab appears to reduce the frequency of rejection at six months by about one-third. However, no statistically significant improvement is seen in the severity of rejection, although the second of the two studies suggests some reduction in the number of patients who experience multiple rejection episodes after twelve months. A statistically significant reduction in the frequency of rejection also persists after twelve months among basiliximab-treated patients, and no additional, adverse events are known to be associated with basiliximab treatment. Although basiliximab and daclizumab both result in 30-40% reduction in acute rejection rates at six months (based on over 1200 renal allograft recipients), their effects on infection, malignancy, chronic rejection, and patient survival remain undetermined.
CONCLUSION
Without question, biopharmaceuticals have come of age, and biologic-based anti-cytokine therapies in particular are changing the practice of medicine. The power of biopharmaceuticals now reaches far beyond acute life-threatening indications, and extends to patients with chronic debilitating conditions such as RA and CD. Importantly, the new generation of biologic therapeutics alters the progression of these disorders, a clinical outcome that has not been achieved with small-molecule therapies. With efficacy established, we are now left to learn just how widespread the use of these agents will become. Will they continue to be limited to severe disorders that are poorly treated with small-molecule drugs? Or will the use of anti-cytokine biopharmaceuticals become a part of first-line medical practice?
Several inherent characteristics provide biopharmaceuticals—particularly monoclonal antibodies and fusion proteins—with important therapeutic advantages. Their selectivity reduces the incidence of side effects, and they tend to have prolonged stability and predictable pharmacokinetics. Moreover, because they are not substrates for cytochrome p450 enzymes, biopharmaceuticals do not alter the metabolism or pharmacokinetics of xenobiotics.
Counterbalancing the advantages of biopharmaceuticals are two general characteristics that constrain their use. First, biopharmaceuticals must be administered parenterally. Second, and more importantly, production costs for these agents are high. In our view, resolution of these issues will greatly accelerate the uptake of biopharmaceuticals into standard medical practice, and permit these agents to fulfill their enormous promise. With a host of new agents in clinical development (Table 1⇑), additional advances are sure to be realized.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Don E. Griswold, PhD, (left to right) is Senior Director of Immunobiology; Xiao-yu R.Song, PhD, is Principle Research Scientist in Immunobiology; David Shealy, PhD, is Associate Director of Protein Biochemistry; and Theodore J. Torphy, PhD, is VicePresident of Research at Centocor.

