CHK1 Inhibitors in Combination Chemotherapy
Thinking Beyond the Cell Cycle
- 1 Department of Neurosurgery, Virginia Commonwealth University, Massey Cancer Center, 401 College Street, Richmond, VA 23298-0035
- 2 Department of Human and Molecular Genetics, Virginia Commonwealth University, Massey Cancer Center, 401 College Street, Richmond, VA 23298-0035
- 3 Department of Medicine, Virginia Commonwealth University, Massey Cancer Center, 401 College Street, Richmond, VA 23298-0035
- 4 Virginia Institute of Molecule Medicine, Virginia Commonwealth University, Massey Cancer Center, 401 College Street, Richmond, VA 23298-0035
Abstract
Cellular sensing of DNA damage, along with concomitant cell cycle arrest, is mediated by a great many proteins and enzymes. One focus of pharmaceutical development has been the inhibition of DNA damage signaling, and checkpoint kinases (Chks) in particular, as a means to sensitize proliferating tumor cells to chemotherapies that damage DNA. 7-Hydroxystaurosporine, or UCN-01, is a clinically relevant and well-studied kinase activity inhibitor that exerts chemosensitizing effects by inhibition of Chk1, and a multitude of Chk1 inhibitors have entered development. Clinical development of UCN-01 has overcome many initial obstacles, but the drug has nevertheless failed to show a high level of clinical activity when combined with chemotherapeutic agents. One very likely reason for the lack of clinical efficacy of Chk1 inhibitors may be that the inhibition of Chk1 causes the compensatory activation of ATM and ERK1/2 pathways. Indeed, inhibition of many enzyme activities, not necessarily components of cell cycle regulation, may block Chk1 inhibitor–induced ERK1/2 activation and enhance the toxicity of Chk1 inhibitors. This review examines the rationally hypothesized actions of Chk1 inhibitors as cell cycle modulatory drugs as well as the impact of Chk1 inhibition upon other cell survival signaling pathways. An understanding of Chk1 inhibition in multiple signaling contexts will be essential to the therapeutic development of Chk1 inhibitors.
Introduction
DNA damage is a ubiquitous process, occurring in all cells and organelles that contain DNA. Damage to DNA can result from errors made during DNA replication as well as from agents that chemically modify or intercalate within the DNA structure. In tumor cells, elevated levels of endogenous reactive oxygen species constantly cause DNA damage, which is one probable factor for the genomic instability of such cells (1, 2). Cells have multiple mechanisms for sensing DNA damage and for initiating processes to permit DNA repair, with varying levels of fidelity. If levels of damage sustained cannot be repaired, the cell may undergo rapid or delayed forms of reproductive cell death (3, 4).
Cellular sensing of DNA damage is mediated through formation of several distinct complexes of proteins, determined by the nature of the DNA lesions, that act to catalyze repair (5, 6). Simultaneously, with the formation of repair complexes, signaling processes are initiated that lead to cell cycle arrest, thereby permitting DNA repair without the occurrence of additional DNA replication or cell division. The sensing of DNA damage with concomitant cell cycle arrest is mediated by pathways involving multiple enzyme activities (Figure 1), including: 1) poly(ADP–ribose) polymerase 1 (PARP1); 2) distinct members of the phosphatidylinositol 3-kinase protein family, known as ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR); 3) checkpoint kinases 1 and 2 (CHK1 and CHK2); 4) the dual-specificity protein phosphatases CDC25A–C; and 5) cyclin-dependent kinases (CDKs, specifically, CDK1 and CDK2/4) (7–16). Additional regulators of these pathways include p53 and the cyclin kinase inhibitor p21. Inhibition of cyclin-dependent kinases pursuant to DNA damage is of central importance for reducing the rate of progression through the cell cycle so that DNA repair can be effected.
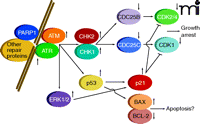
Damage is sensed and repaired in multi-protein complexes. Signaling caused by this damage results in cell cycle arrest and a choice between repair or progression to apoptosis.
A variety of clinical and laboratory observations have led to our understanding of the signaling pathways related to DNA repair. It was recognized that DNA isolated from ataxia-telangiectasia (AT) patients exhibited inherently more evidence of damage and that AT patients were more likely to develop malignancies; the expression of ATM was eventually linked to the disease (17, 18). A multitude of studies in which DNA repair was inhibited elucidated the regulatory pathways downstream of ATM and the related ATR protein; in turn, pharmaceutical companies and ultimately the National Cancer Institute began to explore the development and characterization of novel compounds to inhibit the kinase activities of ATM, ATR, Chk1, and Chk2 [see (19, 20)]. Based on this work, abrogation of DNA damage–induced cell cycle arrest became a major focus of anticancer chemotherapeutic research. Specifically, it was reasoned that chemotherapies that cause DNA damage might be made more effective in the presence of agents that interfere with cell cycle control. By causing “inappropriate” cell cycle progression in tumor cells, frequently characterized by damaged DNA, it was hypothesized, patient survival might be improved. Inhibitors of cell cycle control were thus envisaged as chemosensitizers, exploiting the proliferative nature of transformed cells, and invoking various forms of short-term and long-term reproductive cell death. In this review, the development of Chk1 inhibitors and the cellular responses to such inhibitors are discussed.
The Chk1 Inhibitor 7-Hydroxystaurosporine (UNC-01)
There are almost 400 studies referenced in the National Library of Medicine that cover the use of UCN-01, originally isolated from Streptomyces (21), to explore tumor cell signaling and cell death responses. Although UCN-01 became widely recognized as a broad-spectrum inhibitor of the protein kinase C (PKC) family of enzymes, it proved unique among PKC inhibitors for promoting the activation of Cdk1and Cdk2 and thereby driving cell cycle progression and killing tumor cells (22). More specifically, UCN-01 was demonstrated to abrogate the DNA damage–dependent G2 checkpoint that can be induced by cisplatin treatment; the activity of UCN-01 as a G2-checkpoint inhibitor was found to enhance cisplatin toxicity by as much as sixtyfold (23). Subsequently, several interesting activities associated with UCN-01 were determined, including: 1) radiosensitization associated with the abrogation of ionizing radiation–induced G2/M arrest; 2) enhancement of the toxicity of 1-[beta-D-arabinofuranosyl] cytosine (Ara-C); and 3) the potentiation of lethality of topoisomerase inhibitors, thymidylate synthase inhibitors, and temozolomide (24–28). The cell cycle regulatory effects of UCN-01 were clearly linked to its inhibition of Chk1 and to dysregulation of the dual-specificity phosphatase Cdc25C (29). Although UCN-01 has more recently been shown to inhibit PDK-1 (i.e., the kinase upstream of AKT within the “classic” PI3K pathway), checkpoint abrogation appears to play the major role in the anticancer properties of UCN-01 (30).
In light of its inherent toxicity, as well as its chemosensitizing effects in vitro and in a wide variety of in vivo systems, UCN-01 was chosen by the National Cancer Institute for evaluation in patients. Its pharmacokinetic properties in animal studies had predicted that UCN-01 would lead to sustainable free drug levels in patients and single-agent antitumor effects; however, several phase 1 trials established that the drug had a very long half-life, owing to its binding to alpha-1 acidic glycoprotein, and the levels of free drug in patient plasma were only about 100 nM (31, 32). Although a UCN-01 administration schedule has recently been developed that results in more favorable pharmacokinetics (33), the drug has not shown significant clinical activity, alone or in combination with other agents, and it presently remains unclear whether UCN-01 will become an FDA-approved anticancer drug.
Other Chk1 Inhibitors
Based on the realization that Chk1 was a druggable target and that its inhibition could enhance tumor cell killing by DNA-damaging agents, Chk1 inhibitors distinct from UCN-01 have been investigated. AZD7762 (Astra Zeneca) is a potent Chk1 inhibitor that enhances the toxicity of a variety of DNA-damaging agents against cancer in preclinical models (34). AZD7762 is presently undergoing phase 1 evaluation in a variety of tumor types in combination with DNA-damaging agents. Similarly, PF-477736 (Pfizer), SCH900776 (Schering Plough), and LY2606368 (Eli Lily) have been reported to inhibit Chk1 in tumor cells and enhance chemotherapy sensitivity; they, too, are undergoing phase 1 evaluation in patients (35–37).
Newton’s Third Law and the Development of Combinatorial Cancer Therapeutics
For every action there is an equal and opposite reaction. Cells respond proportionally to stress by adjusting their signaling pathway activities. This observation extends to tumor cells that are exposed, within limits, to a chemotherapeutic agent (e.g., at concentrations that result in a loss of ~30% or less of reproductive survival). Some stress-induced signals are toxic to the cell, depending on the mode of action of the therapeutic agent; other signals, however, may be induced to modulate these stress-induced toxic signals (38). For example, when a carcinoma cell is exposed to ionizing radiation, the epidermal growth factor receptor (EGFR) is activated (39). EGFR activation can, alongside ceramide and reactive oxygen and nitrogen species, play a supporting role in propagating a toxic signal (e.g., activation of the JNK pathway or CD95 death receptor) and coincidentally play a direct role in activation of protective pathways (e.g., activation of AKT and ERK) (40). Inhibitors of EGFR can (like inhibitors of PI3K or MEK1/2) thus radiosensitize tumor cells by shifting the balance of toxic and protective signals toward toxicity (Figure 2) (41). Moreover, toxic signals in this context can offer a chemo-therapeutic advantage because signaling pathway activity levels are usually elevated and more unstable in cancer cells, relative to nontransformed cells. However, the reaction of a tumor cell to a given kinase inhibitor at a concentration that alone causes little cell killing will also be a function of any other administered agent that acts (e.g., in a compensatory fashion) within the nexus of signaling pathways (42).
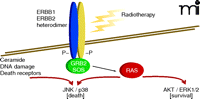
Activated signaling pathways act in balance and can promote both cell survival as well as cell death. The combination of inhibitors that block AKT/ERK1/2 activation with radiotherapy enhances activation of the toxic JNK/p38 pathways to cause tumor cell death.
Chk1 Inhibitors and ERK1/2 Signaling
Almost a decade ago, it was noted that treatment of tumor cells at modest but clinically relevant concentrations of UCN-01 caused a rapid and sustained activation of the ERK1/2 pathway (43, 44). More recently, ERK1/2 activation has been observed in response to the chemically unrelated Chk1 inhibitor AZD7762; similar activation is promoted upon transient expression of a dominant–negative form of Chk1 (45, 46). Chk1 inhibitor–mediated activation of ERK1/2, however, becomes limited upon expression of dominant–negative Chk1. In addition, although knockdown of Chk1 reduces basal ERK1/2 activation, treatment of knockdown cells with Chk1 inhibitors raises ERK1/2 activity (45, 46). In developing Drosophila pupae, loss of Chk1 function has been shown to promote MEK1/2 activation, which provides independent genetic confirmation of studies in cancer cells (47).
In agreement with the concept that ERK1/2 activation is a compensatory protective signal in response to the loss of cell cycle control that occurs upon Chk1 inhibition, tumor cell death rates significantly increase when Chk1 inhibitor–treated cells are subjected to inhibition of MEK1/2 (43–46, 48–61). In addition, the prevention of Ras activity at the cell membrane (i.e., by preventing Ras prenylation; see Figure 3) enhances Chk1 inhibitor–induced toxicity (54, 56, 58, 60). Suppression of mTOR signaling, NFκ B signaling, and HSP90 function has also been shown to enhance, to some degree, Chk1 inhibitor–induced lethality (51, 52, 55). More recently, inhibition of SRC kinase signaling was shown to block Chk1 inhibitor–induced ERK1/2 activation and to strongly enhance Chk1 inhibitor–induced lethality in vitro and in vivo (48, 62).
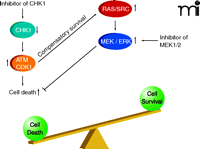
Activated ATM and CDK1 can promote cell killing which is suppressed by a compensatory survival signal through ERK1/2. Inhibition of this ERK1/2 survival signal shifts the balance of signaling causing high levels of tumor cell killing.
Mechanisms of Chk1 Inhibitor–induced ERK1/2 Activation and Tumor Cell Death
Data from several groups have shown that inhibition of checkpoint kinases causes a compensatory activation of ATM [e.g., (45)]. Hypothetically, the compensatory activation of ATM could reflect the enhancement of DNA damage by checkpoint inhibition and concomitant G2/M cell cycle progression, but this hypothesis is challenged by in vitro data using primary human multiple myeloma blasts, which are to all intents and purposes suspended in the G0 phase of the cell cycle. Specifically, even in these “G0-phase cells,” Chk1 inhibitors still activate ERK1/2, and MEK1/2 inhibitors still promote Chk1 inhibitor–mediated toxicity (46, 48, 49, 61). Thus, Chk1 inhibitors may invoke the activation of Cdk1 or other signaling events, rather than acceleration through the cell cycle per se, to promote the activation of ATM. Indeed, the activation of ERK1/2 in response to Cdk1 activation has been established in Xonopus oocytes [see (63)]. Nevertheless, in time course studies, Chk1 inhibitor–induced MEK1/2 phosphorylation was found to precede Cdk1 activation (i.e., dephosphorylation of tyrosine Y15), whereas ectopic expression of Cdk1 or siRNA-mediated Cdk1 knockdown did not alter Chk1 inhibitor–induced ERK1/2 activation (46, 61). Thus, inappropriate activation of Cdk1 cannot be presumed to be the direct outcome of the Chk1 inhibitor–induced activation of ERK1/2. The possibility that Chk1 has alternative targets, which influence ATM and ERK1/2 activity, has not been adequately explored. In contrast, several studies have causally linked DNA damage–induced activation of ATM to DNA damage–induced activation of ERK1/2 (64, 65). In addition, ERK1/2 activation has also been shown to reinforce further ATM activation, suggesting that there is a regulatory feed-forward loop between ATM and the ERK1/2 pathway (65).
Given that Chk1 inhibitors activate ERK1/2 rapidly and do so in a p53-independent fashion, signal transduction between ATM and ERK1/2 is unlikely to require transcription (43, 44). Otherwise, several hypothetical mechanisms by which ATM might promote the SRC-RAS-ERK1/2 pathway might be considered. For example, because Chk1 inhibitors such as UCN-01 and AZD7762 do not inhibit Chk2, Chk1 inhibitor–mediated activation of ATR could allow for activation of Chk2 (66). Chk2–dependent phosphorylation of Cdc25A, in turn, which is a protein phosphatase that is rapidly degraded upon its own phosphorylation, could hinder dephosphorylation of activated Raf-1, thereby enhancing, through RAS and SRC, the ERK1/2 pathway (67–70).
The complex regulation of Cdc25C, which is essential to cyclin B/Cdk1 activity, may also play a role in drug toxicity. Although Cdk1 activation is not a direct result of the ERK1/2 activation that arises from Chk1 and MEK1/2 inhibition, it should be recalled that a dominant–negative form of Cdk1 was found to suppress the toxicity associated with combined Chk1 and MEK1/2 inhibition (46). Inappropriate activation of cyclin-dependent kinases has been noted in multiple systems to kill transformed cells [see (71, 72)]. ERK1/2 pathway signaling promotes Cdc25C phosphorylation at multiple residues so as to increase the phosphatase activity that activates Cdk1 (73). Perhaps more importantly, Chk1-dependent phosphorylation of Cdc25C (at S216) promotes 14-3-3 protein binding and inactivates phosphatase function, thereby increasing phosphorylation of Cdk1 tyrosine residues and reducing Cdk1 activity (74). However, ERK1/2-dependent phosphorylation also causes proteolytic degradation of Cdc25A and Cdc25C, which would thereby facilitate Cdk1 phosphorylation (75). MCL-1, a substrate of Cdk1, is destabilized upon phosphorylation (76); the protective functions of BCL-2 and BCL-XL are similarly inactivated by Cdk1 (77). Accordingly, sustained activation of Cdk1 has been shown to promote apoptosis (78, 79).
In addition to altering ERK1/2 pathway activity, other MAP kinase pathways play a role in regulating Chk1 inhibitor toxicity. For example, the toxicity associated with combining Chk1 inhibitors and ERK1/2 pathway inhibitors has been linked to activation of the JNK and p38 MAPK pathways, and as JNK and p38 are also both downstream targets of ATM, it is probable that ATM signaling could be responsible for the survival and cell death signals caused by drug combination treatment (43–61).
Enhancement of Chk1 inhibitors Lethality by PARP1 Inhibitors
As stated earlier, DNA repair processes involve the formation of multi-protein complexes that associate with damaged DNA. The mechanisms by which DNA damage induces activation of ATM are still not fully understood, but PARP1 is clearly involved [see (7, 80)]. Upon its activation by DNA damage, PARP1 catalyzes the ADP ribosylation of multiple repair proteins (including PARP1 itself), which likely facilitates complex formation. It is of interest, based on prior statements with respect to ionizing radiation–induced ERK1/2 activation and the interconnected nature of ATM and ERK1/2 signaling, that some studies have shown radiation-induced activation of ERK1/2 is dependent on PARP1 function (81). As Chk1 inhibitor–induced activation of ERK1/2 is mediated through an ATM-dependent pathway, it could be hypothesized that inhibition of PARP1 signaling would block any form of ATM activation, and in the case of cells being treated with a Chk1 inhibitor, thereby suppressing the compensatory survival signal of ERK1/2. And indeed, multiple PARP1 inhibitors, as well as knockdown of PARP1 expression, block Chk1 inhibitor–induced activation of both ATM and ERK1/2, and PARP1 inhibitors significantly enhance Chk1 inhibitor lethality in breast cancer cells (Figure 4) (45). However, unlike studies combining Chk1 inhibitors and MEK1/2 inhibitors, where expression of an activated form of MEK1 almost abolishes killing, expression of activated AKT to a greater extent than expression of activated MEK1 is required to reduce PARP1 inhibitor and Chk1 inhibitor lethality. The subtle differences in survival signaling owing to use of MEK1/2 inhibitors versus PARP1 inhibitors in combination with Chk1 inhibitors have yet to be fully explored.
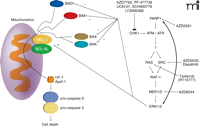
Multiple CHK1 inhibitors promote compensatory activation of the RAS-SRC / MEK1/2 / ERK1/2 pathway. The survival pathway can be inhibited at various points along its course that reveals the true toxicity of CHK1 inhibitor exposure. Killing is mediated via the intrinsic / mitochondrial cell death pathway.
Conclusions
The development of new anticancer drugs, through the clinic to ultimate FDA approval, involves not only the initial assessment of dose-limiting drug toxicity but also the identification of patient populations in which the drug, as a single agent, elicits antitumor responses. As a single agent, it would be predicted that any Chk1 inhibitor would have little to no antitumor effect; instead, Chk1 inhibitors would be expected to enhance the toxicity of drugs that cause DNA damage. Thus, “modulator” drugs such as Chk1 inhibitors are placed at a relative disadvantage in the drug development pipeline. For example, and in a similar manner to Chk1 inhibitors, Cdk inhibitory drugs, such as flavopiridol (Alvocidib), which act to suppress Cdk7/Cdk9 activity, have not shown significant anticancer effects as single agents but are showing some ability at reversing bortezomib (Velcade) resistance in blood tumor cells. In our own studies, in breast cancer tumor xenografts, a two-day exposure to either UCN-01 or a MEK1/2 inhibitor did not have any impact on tumor growth, whereas combined exposure to both drugs could be shown nearly to eliminate tumor growth (57). Collectively, these findings suggest that the “standard” model of drug development needs to be slightly modified, taking into account new advances in our understanding of how specific combinations of kinase inhibitors, although individually ineffective, can lead to high levels of tumor cell killing.
Nevertheless, despite the lack of single agent antitumor effects, the NCI has invested considerable resources into the clinical development of UCN-01 as a modulator of cell cycle checkpoints with the possible goal of enhancing the toxicity of marketed DNA-damaging anticancer drugs. Unfortunately, in the phase 1 and phase 2 studies that have been and are being performed in patients wherein Chk1 inhibitors are combined with “traditional” cytotoxic chemotherapeutic drugs, there have as yet been no widespread reports in the literature of profound enhancements in tumor control or patient survival.
One relatively unexplored component of Chk1 biology in the development and application of Chk1 inhibitors has been their effect on activation of ATM and of the ERK1/2 pathway, as discussed above. As activation of ERK1/2 is frequently associated with tumor cell survival, it may well be responsible for reducing the overall chemosensitization effect caused by checkpoint abrogation. For example, elevated levels of ERK1/2 signaling have often been associated with protection of tumor cells from the toxic actions of therapeutic drugs and ionizing radiation (82–85). Thus the rational therapeutic application of Chk1 inhibitors may require, in addition to combination with the “traditional” cytotoxic agent, the concurrent inhibition of compensatory ATM or ERK1/2 activation (e.g., through the use of PARP1 or MEK1/2 inhibitors, respectively). Such combined inhibitor therapy is under investigation, for example, in the case of EGFR and VEGFR inhibitors concurrent with gemcitabine treatment (86).
Acknowledgements
This work was funded by the Public Health Service (Grants R01-DK52825, P01-CA104177, R01-CA108325, R01-CA141703, R01-CA150214, R01-CA100866, and R01-CA93738) and by the U.S. Department of Defense (Grants DAMD17-03-1-0262 and W81XWH-10-1-0009). The work was also funded by The Jim Valvano “Jimmy V” Foundation. PD is the holder of the Universal Inc. Professorship in Signal Transduction Research; S.G. is the holder of the Olsen Distinguished Professorship; PBF holds the Thelma Newmeyer Corman Chair in Cancer Research in the VCU Massey Cancer Center and is an SWCRF Investigator.
Footnotes
-
Authorship contribution.
Wrote or contributed to the writing of the manuscript: Dent, Grant, Fisher, and Dai.
-
Other: Tang and Yacoub performed literature searches; Grant, Fisher, and Dai proofread the manuscript and contributed additional concepts.
- Copyright © 2011
References
Paul Dent, PhD, is Professor and Vice Chair of Research in the Department of Neurosurgery at Virginia Commonwealth University. Email pdent{at}vcu.edu; fax 804-827-1014.


