STAT3 SIGNALING: Anticancer Strategies and Challenges
Abstract
Multiple lines of evidence place STAT3 at a central node in the development, progression, and maintenance of many human tumors, and STAT3 has been validated as an anti-cancer target in several contexts. STAT3 modulates the transcription of a variety of genes involved in the regulation of critical functions, including cell differentiation, proliferation, apoptosis, angiogenesis, metastasis, and immune responses. For many cancers, elevated levels of activated STAT3 have been associated with a poor prognosis. We review approaches that have been pursued to target STAT3, and we highlight some of the promises and challenges associated with developing an anticancer drug that might therapeutically inhibit the STAT3 signaling pathway.
Introduction
Signal transducer and activator of transcription 3 (STAT3) is a member of a family of seven proteins (STATs 1, 2, 3, 4, 5a, 5b, and 6) that relay signals from activated cytokine and growth factor receptors in the plasma membrane to the nucleus, where they regulate gene transcription (1–3). STAT3 modulates the transcription of responsive genes involved in the regulation of a variety of critical functions, including cell differentiation, proliferation, apoptosis, angiogenesis, metastasis, and immune responses (2, 4–6). Multiple lines of evidence place STAT3 at a central node in the development, progression, and maintenance of many human tumors, validating STAT3 as an anticancer target (Table 1) (2, 4–7). Cellular transformation by the viral oncogene v-src requires activated STAT3 (2, 7–10), and the transfection and expression of a constitutively activated form of STAT3 is sufficient to transform immortalized fibroblasts and normal epithelial cells derived from breast and prostate tissue (8). Most of the major human malignancies manifest elevated levels of constitutively activated STAT3 (Table 1) as well as transcriptional profiles that are consistent with STAT3-regulated gene expression (2, 4, 6, 7). For many cancers, elevated levels of activated STAT3 have been associated with a poor prognosis (Table 1) (2, 4, 5, 11). STAT3-activated genes block apoptosis, favor cell proliferation and survival, promote angiogenesis and metastasis, and inhibit antitumor immune responses (2, 4–6, 12). Tumor cell lines bearing constitutively activated STAT3 require continued STAT3 activation, a phenotype that has been termed “oncogene addiction” (4). In contrast, approaches that disrupt STAT3 signaling lead to growth inhibition and apoptosis in tumor cell lines and can impair tumor growth in mouse xenograft cancer models (Table 1) (2, 5, 6, 9, 13–19). Although knockout of STAT3 leads to embryonic lethality in mice, the cumulative data in postnatal mice (conditional knockouts) indicates that STAT3 may be dispensable for the function of normal cells and tissues (2, 4). The ability of nontransformed cells to withstand inhibition of STAT3 signaling suggests that a potential anticancer drug that targets STAT3 might be well tolerated (2, 4).
STAT3 in the Context of Various Cancers: Validation as an Anticancer Target
STAT3 and STAT1 have great sequence similarity and function as transcription factors that bind to very similar DNA sequences, but in many physiological contexts, they are regulated in a reciprocal manner to produce opposing effects (12, 17, 20). In the cancer context, activated STAT3 is oncogenic, whereas activated STAT1 behaves as a tumor suppressor (12). Therefore, the selective inhibition of STAT3, without interference in STAT1 signaling, might be proposed as the basis for anti-cancer drug development. However, in experimental situations, STAT1 and STAT3 may be functionally interchangeable in promoting cytokine-induced programs of gene expression [e.g., STAT1 on its own may affect such gene expression via the interleukin-6 receptor, whereas STAT3 may operate via the interferon-gamma receptor (2, 12)]. It should also be noted that constitutively activated STAT5, which shows little sequence similarity with STAT3, has also been implicated in oncogenesis (2, 21). Here, we review approaches that have been pursued to target STAT3, and we highlight some of the challenges associated with developing an anticancer drug that might therapeutically inhibit the STAT3 signaling pathway.
STAT3 Domain Structure, Signaling, and Regulation
STAT3 is structurally typical of the STAT family: an N-terminal coiled-coiled domain involved in protein–protein interactions; a DNA binding domain; a Src homology-2 (SH-2) domain; and a C-terminal transactivation domain (5, 22). In the canonical STAT3 signaling pathway (Figure 1), activation of cell surface receptors by growth factors and cytokines induces the specific phosphorylation of receptor tyrosine residues to create docking sites for the recruitment of latent cytoplasmic STAT3 (1–3, 5, 22). STAT3 docking to receptor phosphotyrosine (pY) sites is mediated through its SH-2 domain. The binding of STAT3 at receptor pY sites leads to the phosphorylation of a specific tyrosine residue of STAT3 (Y705) in the C-terminal domain, and this phosphorylation activates STAT3 (1–3, 5, 22). The kinase activity that phosphorylates STAT3 at Y705 is determined by the specific receptor pY binding site with which STAT3 interacts. Growth factor receptors, with their intrinsic receptor tyrosine kinase (RTK) activity, phosphorylate STAT3; activated cytokine receptors, in contrast, associate with a Janus kinase (JAK) that phosphorylates STAT3 (1, 3, 22). The phosphorylation of STAT3, in turn, promotes homodimerization, wherein the SH-2 domain of each monomer interacts with the Y705 residue of its partner. STAT3 dimers translocate to the nucleus, where they bind to specific DNA response elements in the promoter regions of responsive target genes to regulate their transcription (Figure 1) (1–3, 5, 22). Phosphorylation of a single serine (S727) in the C-terminal transactivation domain of STAT3 allows for the maximal activation of transcription of responsive genes (4, 22).
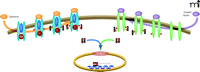
In the canonical STAT3 signaling pathway, activation of cell surface growth factor and cytokine receptors induces specific tyrosine phosphorylation (small red circles) of receptor chains to create docking sites for the recruitment of latent cytoplasmic STAT3 (dark gray geometries), which contain SH-2 domains (shown as indentations) that recognize sites of tyrosine phosphorylation. After the recruitment of STAT3 to activated receptors, STAT3 becomes activated by phosphorylation (at Y705; see text for details). Phosphorylation of STAT3 can be catalyzed by the intrinsic tyrosine kinase activity of the activated growth factor receptor (green elongated ovals) or by the Janus kinase (JAK) that associates with activated cytokine receptors (blue elongated ovals). Phosphorylated STAT3 homodimerizes, via reciprocal intermolecular interactions between SH-2 domains and phosphorylation sites, and dimers translocate into the nucleus. Passage of dimers through the nuclear pore complex (red rings) is facilitated through interactions with importin α5, importin α7, and importin β; nuclear import of nonphosphorylated STAT3 may also occur by importin α3 (not shown). In the nucleus, STAT3 dimers bind to promoter elements of responsive target genes to regulate their transcription. See Tables 1 and 2 for specific activators and targets of STAT3 activity.
Under normal conditions, both the amplitude and duration of receptor-induced STAT3 activation are tightly controlled by a variety of endogenous protein regulators (1, 2, 22–26). For example, several protein tyrosine phosphatases (PTPs) have been implicated in the termination of STAT3 signaling, including SHP-2, PTP1B, PTPɛC, TC45, and SHP-1 (1, 22, 25, 26). SHP-2 is ubiquitously expressed, and like SHP-1, contains two N-terminal SH-2 domains and a C-terminal PTP domain (1, 22, 25). SHP-2, by virtue of its two SH-2 domains, associates with pY-sites of growth factor receptors, cytokine receptors, gp130, JAKs, members of the Src family of tyrosine kinases (SFKs), and STAT3. SHP-2 may in turn become phosphorylated by RTK activities, JAK1, JAK2, and SFKs (1, 22, 25). All of the pY components of STAT3 signaling pathways are potential substrates for PTPs (1, 22, 25). Because only STAT3 dimers bind to DNA, the nuclear PTP TC45 may be important in the termination of STAT3-mediated transcriptional activation (22, 26).
The constitutively expressed protein inhibitor of activated STAT (PIAS) family members share a highly conserved domain structure (2, 22, 24, 25). The N terminus includes a domain (i.e., the SAP domain) that can bind to AT-rich DNA sequences [i.e., scaffold/matrix attachment regions (S/MARs)] that are frequently found in nearby gene enhancers; this N-terminal SAP domain also contains an LXXLL motif, implicated in interactions between nuclear receptors and their coregulators (24, 25). Next to the SAP domain within the PIAS protein sequence is a Pro-Ile-Asn-Ile-The (PINIT) motif, which is involved in the nuclear retention of PIAS proteins. Midway within the PIAS sequence is a RING finger-like zinc binding domain (RLD) C-terminal to which is a highly acidic domain (AD) that contains a SUMO1 interaction (SIM) motif (24, 25). At the C terminus, there is a conserved Ser/Thr-rich (S/T) region. PIAS3 binds to STAT3 specifically and blocks its DNA binding activity and transcriptional activation (2, 22, 24, 25). The SUMO E3 ligase activity of PIAS3 promotes the sumoylation of a number of proteins, including STAT3, but it is unclear whether sumoylation contributes to the inhibition of STAT3 function (24, 25).
The family of suppressors of cytokine signaling (i.e., SOCS1–7 and CIS) contain a central SH-2 domain flanked by a forty-residue C-terminal SOCS box domain and an N-terminal domain of variable length (2, 22, 23, 25). The SOCS SH-2 domain mediates binding to specific pY-containing motifs in JAKs, gp130, and cytokine receptor chains (22, 23, 25). The SOCS box domain contains three alpha-helices bound to an E3 ubiquitin ligase complex that can promote the ubiquitination and proteasomal degradation of SOCS binding partners (22, 23, 25). SOCS proteins behave as classical feedback inhibitors, induced by the cytokine-mediated activation of the JAK/STAT pathway that they function to inhibit (22, 23, 25). Furthermore, the transcription of SOCS3 is activated by STAT3. SOCS3 can block STAT3 signaling by any of three mechanisms: by direct binding and inhibition of JAKs, by competing with STAT3 for pY-binding sites on activated receptor chains, or by binding signaling proteins and targeting them for proteasomal degradation (2, 22, 23, 25). In addition, the expression of SOC3 can also be upregulated in a STAT3-independent mechanism that involves elevated intracellular cAMP levels that activate protein kinase A (PKA) (27–29). For example, G protein–coupled receptors (GPCRs) that signal through Gs proteins (e.g., PGE2, β-adrenergic, and prostacyclin receptors) produce increased levels of cAMP, which activate PKA and trigger the accumulation of SOC3, thereby inhibiting interleukin-(IL)-6–induced STAT3 activation (27–29).
Mechanisms of STAT3 Activation in Tumor Cells
Although increased levels of phosphorylated STAT3 have been detected in the majority of human cancers and tumor-derived cell lines, no naturally occurring mutations that produce constitutive activation of STAT3 have yet been detected (2). There are, however, multiple potential upstream inputs that lead to the activation of STAT3, including: the human epidermal growth factor receptor (EGFR), a member of the ErbB/HER family of RTKs; the family of IL-6–type (IL-6) cytokine receptors that form complexes with gp130 and JAKS; and several GPCRs (Figure 1) (1, 3, 7, 9, 10, 22, 30, 31). Multiple growth factors (e.g., EGF, TGFα, PDGF, and CSF1) and cytokines (e.g., IL-6, LIF, CT-1, CNTF, IL-10, IL-11, and OSM) have been shown to activate STAT3 (1–3, 9, 20, 22). In addition, SFKs (e.g., Src, Lck, Hck, Lyn, Fyn, and Fgr) either activate STAT3 directly or do so downstream of the activation of RTKs or GPCRs (9, 10, 30). Elevated levels and/or constitutive activation of signaling components that promote STAT3 phosphorylation have been detected in many tumor types and are frequently associated with a poor prognosis; associated molecular events may include elevated EGFR expression levels, EGFR mutations that result in constitutive RTK activation, overexpression of Src or other SFKs, and mutations that hyperactivate JAKs (2, 3, 9, 10, 23, 32–34). Elevated levels of STAT3-activating ligands, such as TGFα or IL-6, have also been detected in the serum and/or the tumor microenvironment of patients with a variety of human malignancies (2–4). The increased amounts of TGFα or IL-6 that sustain activation of STAT3 in these cases may be produced in an autocrine, paracrine, or endocrine manner (2–4).
In addition, mutations that disrupt the epigenetic control (e.g., hypermethylation of gene sequences) of endogenous regulators of STAT3 signaling have also been identified in a variety of human malignancies (2, 4, 22, 23, 25). Inactivation or reduced expression of SOCS, PIAS, and PTP proteins may also lead to a sustained activation of STAT3 signaling in tumorigenesis. Although the list of potential inputs that may contribute to cancer-associated STAT3 activation is daunting, the perceived benefits of a targeted STAT3 anticancer drug have prompted many efforts to pursue this goal.
Strategies to Prevent STAT3 Activation
One approach to prevent STAT3 from promoting transcription would be to inhibit the tyrosine kinase activities of growth factor receptors, JAKs, and intracellular SFKs that activate STAT3; however, in most cancers, the inhibition of multiple kinase pathways is impractical (Table 2) (2, 3, 9, 13, 31–33, 35). A related strategy would be to antagonize those growth factors and cytokines detected in the serum and/or the tumor microenvironment of patients (Table 2) (2, 3, 9).
Strategies and Challenges to Therapeutic Intervention into STAT3 Signaling
Two therapeutic strategies to block EGFRs have been implemented. Monoclonal antibodies (e.g., cetuximab and panitumumab) have been developed to target the extracellular domain of the EGFR, and small molecules (e.g., gefitinib, erlotinib, and lapatinib) have been designed to inhibit the RTK activity of the EGFR (Table 2) (2, 3, 9. 36). Monoclonal antibodies that antagonize ligand–EGFR interactions and disrupt downstream signaling may also induce activation of antitumor cellular immunity (2, 3, 9, 36). The FDA has approved the use of anti-EGFR monoclonal antibodies for treating colorectal cancer and head and neck squamous cell carcinomas (HNSCC) (2, 3, 9). RTK inhibitors have been approved by the FDA for pancreatic cancer and non-small-cell lung cancer (2, 3, 9). Despite the prevalence of high EGFR expression levels associated with various tumors, along with a high incidence of somatic EGFR mutations that constitutively activate the EGFR in tumors, clinical data show that many patients are refractory to EGFR inhibitor treatment (2, 3, 9, 36). Several mechanisms for the development of resistance to EGFR-targeted therapies have been proposed, including secondary mutations of the EGFR that preclude binding to monoclonal antibodies or to small-molecule RTK inhibitors; alternative mutations may result in activation of the Ras pathway, epithelial-mesenchymal transition, or downstream signaling pathways (36).
Upon activation of the IL-6 receptor, the receptor α-subunit recruits two gp130 signal–transducing subunits to the receptor complex; consequently, gp130-associated JAKs (i.e., JAK1, JAK2, and Tyk2) become activated and thereby implement, within the cytoplasmic tail of gp130, pY docking sites for STAT3 (1, 22). Several small-molecule JAK inhibitors (e.g., AG490, LS-104, ICNB18424, and CEP701) have been tested in tumor xenograft models and some clinical trials for cancer (Table 2) (31–33). Both in vitro and in vivo, AG490 inhibits JAK2 activity, reduces activated STAT3 levels, blocks STAT3 DNA binding, and inhibits the growth of leukemic cells (6, 31). LS-104, a closely related analog of AG490, has progressed into phase II clinical trials for acute lymphoblastic leukemia (31). INCB1824, a small-molecule inhibitor of both JAK1 and JAK2, suppresses levels of phosphorylated STAT3 in subjects with wild-type JAK2 or with the gain-of-function V617F JAK2 mutation, as revealed in a phase 1–2 clinical trial of myelofibrosis patients (31, 33). INCB1824 appears to elicit a clinical benefit in myelofibrosis patients while producing a mild myelosuppression adverse response in ten percent of patients. In a phase II study of CEP-701, a JAK2 inhibitor, levels of phosphorylated STAT3 have been reported to decrease in responders while on therapy, although its modest efficacy in myelofibrosis patients is associated with a mild myelosuppression as well as a mild but frequent GI toxicity (32).
Elevated Src levels and/or kinase activity have been reported in a number of cancers, and activation of SFK family members and STAT3 occurs downstream of growth factor RTKs as well as GPCRs (9, 10, 30, 35). Several small-molecule inhibitors of Src and other SFKs are currently in clinical development for a variety of solid tumors (Table 1) (9). Dasatinib inhibits SFK and BCR/ABL and has been approved for use, after imatinib treatment, in patients with chronic myelogenous leukemia and for Philadelphia chromosome–positive acute lymphoblastic leukemia (9). However, there are reports that sustained Src inhibition by Dasatinib only transiently inhibits STAT3 activation (35). Reactivation of STAT3 after prolonged Dasatinib treatment appears to be mediated through altered JAK-STAT binding and JAK kinase activity, perhaps representing a compensatory pathway that allows for cancer survival and proliferation despite chronic Src inhibition (35). XL999, which inhibits a number of growth factor RTKs (e.g., VEGFR, PDGFR, and FGFR) and SFKs is associated with serious cardiovascular toxicities in phase 1 and 2 trials (9).
The relatively modest response rates of tumors to therapeutic agents that target the EGFR, JAKs, and SFKs illustrate that the inhibition of single pathways may not be sufficient to block the activation of STAT3. Indeed, there is evidence that STAT3 contributes to the development of resistance to tyrosine kinase inhibitors.
Strategies to Block Protein–Protein Interactions in STAT3 Signaling
Several groups have synthesized pY-containing peptide sequences to target the STAT3 SH-2 domain as a means of precluding STAT3 recruitment to activated receptors and STAT3 homodimerization (Table 2) (2, 5, 6, 14). The first pY-containing peptide investigated for inhibiting STAT3 activity was based on the synthesis of a peptide sequence that included the Y705 residue that is phosphorylated in the native STAT3 protein to promote homodimerization; in vitro, the synthesized peptide sequence was found to inhibit the binding of STAT3 to DNA (6, 14). This approach has been expanded upon by several groups to include pY-containing peptide sequences from other proteins that interact with STAT3 SH-2 domains (6, 14). Phosphorylation of specific tyrosine residues within the EGFR (i.e., Y1068 and Y1086) is essential to the recruitment of monomeric STAT3 to the activated EGFR (2, 6). A phosphodecapeptide based on the sequence surrounding Y1068 within the EGFR was found to bind to non-phosphorylated STAT3 and to inhibit the DNA binding of phosphorylated STAT3 in vitro (6). Similarly, other pY-containing motifs that interact with the STAT3 SH-2 domain have been investigated; these motifs include the four distinct pY-containing (pYXXQ) motifs within the gp130 signal-transducing subunits of the activated IL-6 receptor complex and multiple pY-containing motifs from cytokine receptor chains [e.g., leukemia inhibitory factor receptor, IL-10 receptor, and granulocyte colony–stimulating factor receptor] (22). Peptide aptamers that inhibit the EGFR-mediated phosphorylation of STAT3 were also identified in a modified yeast two-hybrid screen (2). However, peptide inhibitors have unfortunately been characterized by poor cell permeability and metabolic stability, which has prompted a pursuit of peptidomimetics and small-molecule derivatives (6, 14). Several small-molecule inhibitors have been identified through peptide- and peptidomimetic-inspired rational design including in silico computational approaches and in vitro high-throughput screening (6, 14). Despite advances in identifying peptides and small molecules that target the STAT3 SH-2 domain, molecules with therapeutic anticancer activity have yet to be discovered (6, 14). An alternate strategy to block STAT3–protein interactions employs G-rich oligodeoxynucleotides that form potassium-dependent four-stranded structures (i.e., G quartets) to occupy sites within the STAT3 SH-2 domains (Table 2) (2, 5, 6, 14). Although G quartet oligodeoxynucleotides can disrupt the homodimerization necessary for STAT3 activity, their large size and potassium dependence limit cellular permeability and present significant challenges for in vivo delivery (6).
Strategies to Block Nuclear Translocation of STAT3
The role of activated STAT3 as a DNA-binding transcription factor depends upon the trafficking of homodimers from beneath the plasma membrane and into the nucleus (2, 4–6, 22, 26, 37). Preventing the transit of STAT3 dimers through the nuclear pore complex would thus provide a means to block STAT3 transcriptional activity (Table 2) (26, 37). However, the exact process and identity of the components that mediate STAT3 translocation from the periphery of the cell to the nucleus have yet to be defined. Large protein complexes may enter the nucleus only through the nuclear pore complex, in a process facilitated by importin α and β proteins (26, 37). Importins α5 and α7, in conjunction with importin β, have been implicated in passage of phosphorylated STAT3 through the nuclear pore (26). However, it has also been reported that STAT3 nuclear import may be independent of tyrosine phosphorylation and mediated by importin α3 (37). The nucleocytoplasmic shuttling of STAT3 reflects a dynamic steady state between rates of import and export (26, 37). But intriguingly, phosphorylated STAT3 appears be imported more rapidly than nonactivated STAT3 (26). Within the nucleus, STAT3 is dephosphorylated by the nuclear PTP TC45, thereby becoming a substrate for exportin-1–mediated export (Figure 2) (26). To date, no small-molecule inhibitors of importins α3, α5, or α7 have been identified, and the effects, if any, of a recently identified importin β inhibitor, Karyostatin 1A, have yet to be reported with regard to STAT3 nuclear import (26, 37). Inhibition of exportin 1 by Leptomycin B or Ratjadone A interferes with nuclear export of STAT3; concomitantly, the treatment reduces levels of STAT3 phosphorylation as well as STAT3-mediated transcription and causes cells to undergo apoptosis (26). Despite these interesting results, any small-molecule drug that inhibits general trafficking across the nuclear membrane is likely to be deleterious (Table 2) (26). Whether an inhibitor of nuclear pore transit can be developed with sufficient STAT3 selectivity remains to be determined.
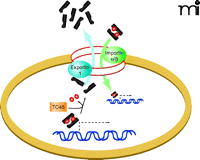
The translocation of STAT3 across the nuclear membrane represents one target of intervention in the STAT3 signaling pathway. Inhibition of exportin-1 (i.e., the ortholog of yeast Crm1) by the fungal toxin Leptomycin B, for example, results in nuclear retention of STAT3, where phosphotyrosine phosphatase activities (e.g., TC45) inactivate STAT3 and preclude its function as a transcription factor. The effects of importin inhibitors upon STAT3 function are not yet defined. (See text for details.)
Strategies to Block STAT3–DNA Binding
Double-stranded oligodeoxynucleotides (dsODN), designed and synthesized to mimic cis regulatory elements within genes that are activated by STAT3, compete for active STAT3 dimers and thereby block induction of STAT3-responsive gene expression and tumor cell growth in vitro (Table 1) (2, 5, 6, 13, 17, 19, 38). The inhibition of STAT3-mediated transcription, elicited by daily intratumoral injections of a STAT3-specific dsODN decoy, has also been reported to reduce tumor cell growth in mouse xenograft models (13, 17, 19, 38). In a safety trial conducted in cynomolgus monkeys, intramuscular injection of 3.2 mg/kg of a STAT3 dsODN decoy produced no observable adverse effects despite reductions in STAT3 target gene expression at the site of injection (38). A phase 0 pharmacodynamic clinical study is underway to test the safety and biological effects of intratumoral injection of a STAT3 dsODN decoy in HNSCC patients. Although preliminary results of this study demonstrate that the STAT3 decoy abrogates the expression of STAT3 target genes in the tumor following a single intralesional injection, the present formulation is rapidly degraded in serum and would not be amenable to systemic administration. Although chemical modification of the decoy may be able to improve metabolic stability of the dsODN, thereby allowing systemic administration, the effective delivery of such modified dsODNs to tumor cells remains a significant challenge (Table 2). Peptide aptamers that inhibit STAT3 DNA binding in vitro were identified in a modified yeast two-hybrid screen, but poor cell permeability and metabolic stability in vivo also hinder the use of peptide-based STAT3 inhibitors (2).
Natural Product Inhibitors
A number of natural products, including guggulsterone, honokiol, curcumin, resveratrol, flavopiridol, and cucurbitacin, have been shown to suppress STAT3 activation and concomitantly inhibit the growth of tumor cell lines (Table 2) (2, 6, 7, 14–16, 18). Several of these natural products have been reported to be of benefit in animal tumor models, either alone or in combination with other antitumor agents (15, 16, 18). However, because these compounds have also been reported to provide a variety of therapeutic benefits unrelated to cancer, the relationship between any therapeutic activity they may possess and STAT3 inhibition remains obscure.
High Throughput Screening
An in vitro high-throughput screen of over 17,000 compounds was conducted in a peptide-binding competition assay. Each tested compound was investigated for its ability to compete for STAT3 binding against a fluorescein-labeled pY-containing oligopeptide derived from the IL-6 receptor signaling subunit gp130 (14). Stattic, one of the first nonpeptide STAT3 inhibitors, was identified in this screen and was found to interfere with the phosphorylation of inactive STAT3 as well as the dimerization and nuclear translocation of activated STAT3 (14). A more extensive HTS campaign of more than 195,000 compounds was recently completed by the Scripps Research Institute Molecular Screening Center as part of the Molecular Library Screening Center Network (MLSCN) effort sponsored by the NIH. The assay utilized a STAT3-dependent luciferase reporter stably transfected into the STAT1-deficient human U3A fibrosarcoma cell line as a background that is deficient in STAT1 (PubChem AIDs 862, 1265 and 1399). Subsequent to HTS results, certain compounds were evaluated in terms of concentration response IC50 values (values are accessible at http://pubchem.ncbi.nlm.nih.gov). Over one hundred compounds were identified that inhibited STAT3-mediated transcription with IC50 values below 10 μM (without significantly affecting STAT1 activity). To date, however, there have been no publications documenting the characterization of the chemical structures and biological activities of the STAT3-selective inhibitors alleged through this high-throughput protocol. High-content fluorescent imaging assays are expected in the near future to identify inhibitors of STAT3 functions from compound libraries.
Conclusions
Despite the validation of STAT3 as a key target involved in human malignancy, a STAT3-selective targeted anticancer drug has after nearly two decades of research yet to be clearly identified. Although no constitutively activating STAT3 mutations have been detected in patient samples, the activation of STAT3 signaling is characteristic of many types of cancer, and elevated STAT3 expression or activation is associated with a poor prognosis. Mutations that affect negative regulators of STAT3 signaling have been identified in a variety of human malignancies, although a direct mechanistic link to STAT3 activation has yet to be reported. Monotherapies, using either anti-EGFR antibodies or small-molecule tyrosine kinase inhibitors to prevent STAT3 activation, show only limited efficacy. Because multiple growth factors, cytokines, GPCR ligands, and SFKs activate STAT3, blockade of any single pathway to STAT3 activation may not sufficiently abrogate STAT3. STAT3 occupies a point of convergence for many signaling pathways, so that the effective targeting of STAT3 will require intervention into multiple pathways. In addition, the significant similarity between STAT1 and STAT3, along with the potential for STAT1–STAT3 heterodimerization, may impede development of STAT3-selective targeting strategies. The STAT3 SH-2 domain is involved in both the recruitment of STAT3 to activated receptors and the formation of transcriptionally active STAT3 dimers. Peptide and peptidomimetic inhibitors have been developed that block these protein–protein interactions, but poor cell permeability and limited metabolic stability are major hurdles in the pursuit of small-molecule derivatives. Although several small-molecule STAT3 inhibitors have been identified through peptide- and peptidomimetic-inspired rational design, including in silico computational approaches and high-throughput screening, molecules with therapeutic anti-cancer activity have yet to be discovered. The pursuit of small molecules that affect STAT3 trafficking is an emerging strategy to block the transcriptional activity of STAT3. Oligonucleotides have shown promise in inhibiting STAT3 function and tumorigenesis, and a phase 0 pharmacodynamic clinical study is underway to test the safety and biological effects of intratumoral injection of an oligonucleotide inhibitor of STAT3 in HNSCC patients. Chemical modifications will be required to improve the metabolic stability of oligonucleotides to allow systemic administration, as will the development of delivery methods. It is anticipated that the combination of innovative STAT3 high-throughput assay formats will identify novel small-molecule leads that may be developed into anticancer drug candidates.
Acknowledgments
This work was supported by the National Institutes of Health [Grant P50CA097190] , the American Cancer Society, and the NExT-CBC Project ID 1015 Agreement Number 29XS131 funded by the National Cancer Institute.
Footnotes
-
Authorship contributions
Wrote or contributed to the writing of the manuscript: Johnston and Grandis.
- Copyright © 2011
References
Paul Johnston, PhD, is Research Associate Professor in the Department of Pharmaceutical Sciences and Manager of the University of Pittsburgh Drug Discovery Institute Screening Facility.

Jennifer R. Grandis, MD, FACS, is an Assistant VP for Research Integration and Professor of Otolaryngology and Pharmacology at the University of Pittsburgh School of Medicine. Address correspondence to JRG. E-mail grandis-jr{at}upmc.edu; fax 412-647-2080.


