Transcription of MDR1
A Therapeutic Target
- Kathleen W. Scotto, Ph.D. and
- Robert A. Johnson
- Memorial Sloan-Kettering Cancer Center and, Weill Graduate School of Cornell University, 1275 York Avenue, New York, NY 10021
- Address correspondence to KWS. E-mail k-scotto{at}mskmail.mskcc.org; fax 212-639-8972.
Abstract
Two decades ago, the overexpression of P-glycoprotein (Pgp) was first demonstrated to mediate the energy-dependent efflux of a variety of chemotherapeutic agents from tumor cells, resulting in the development of multidrug resistance (MDR). Not surprisingly, this discovery triggered an ongoing search for agents that would inhibit Pgp function, with the hope that by doing so the MDR phenotype could be reversed. As our understanding of Pgp function and pharmacokinetics has increased, this quest has become more urgent, as well as more complex.
The sea squirt Ecteinascidia turbinata, shown above as grape-like clusters of sheathed individuals, is the source of a novel chemotherapeutic agent. The organism
produces ET-743, which appears to interfere, in an exquisitely specific manner, with cellular transcription programs involved
in cancer. (Image provided by PharmaMar.)
Introduction
The importance of inhibiting P-glycoprotein (Pgp) function in tumor cells as a potential means of enhancing drug efficacy was advanced more than two decades ago (1, 2). This notion has been augmented by very recent studies suggesting that Pgp plays a general anti-apoptotic role that extends beyond resistance to chemotherapeutics, because tumor cells that overexpress Pgp are resistant to a wide range of caspase-dependent apoptotic inducers including tissue necrosis factor, Fas ligand, serum starvation, and UV irradiation (3–6). On the other hand, it has become clear, particularly from knockout mouse studies (7, 8), that Pgp plays a critical function in normal cells, and that the inhibition of Pgp expression in regions such as the blood brain barrier and colon epithelium is likely to affect drug pharmacokinetics and biodistribution, resulting in increased toxicity of many clinically useful chemotherapeutic agents. Thus, new approaches to circumventing Pgp overexpression, ideally with limited effect on constitutive expression in normal cells, are needed. This review addresses the possibility that Pgp overexpression can be circumvented by inhibiting the transcriptional activation of the MDR1 gene, which encodes Pgp .
Regulation of RNA Polymerase II–Mediated Transcription
Until recently, the probability that agents could be identified that would specifically inhibit a given gene or subset of genes was viewed skeptically (with the notable exception of hormone-regulated gene families). After all, some components of the transcription machinery (e.g., RNA Polymerase II [Pol II] and TATA-binding protein) are required for expression of all protein-encoding genes, and several others (e.g., Sp1 and NF-Y) are involved in the regulation of a very large subset of genes, making it difficult to envision how selectivity could be achieved. However, the past decade of transcription research has made it clear that gene regulation is much more complex than initially suspected. Whether a gene is activated or repressed in a given cell type under a given set of physiological conditions is determined by the presence, complexity, and accessibility of a myriad of response elements within the promoter, as well as by the complement of transcription factors available to interact with these elements (9). Consequently, the interplay of multiple factors is often unique to a given promoter at any given time.
Moreover, superimposed on the protein–DNA interactions that mediate transcriptional output is the influence of a dynamic chromatin ultrastructure. In the past few years, a number of coregulators, proteins that interact with transcription factors and play an intermediary role in the transduction of information from promoter-bound proteins to the basal transcriptional machinery, have been identified. More than half of these molecules interact with chromatin, perturbing its structure in ways that activate or repress transcription. Thus, it appears that most gene regulation is carried out not by a single transcription factor, but by specialized multiprotein complexes that include both DNA-binding proteins and coregulators. These complexes are assembled using both common basic components, as well as unique components that tailor a complex for the transduction of signals initiated by particular developmental or metabolic pathways. It is therefore clear that the first step toward identifying potential targets for modulation of transcription at a given gene is an in-depth understanding of the DNA elements and protein factors that regulate its constitutive and inducible expression.
Constitutive (Uninduced) Transcription of theMDR1 Gene
MDR1 belongs to a group of protein-encoding genes that lack a consensus TATA box within the proximal promoter region. Instead, basal transcription is directed by an initiator (Inr) sequence that encompasses the transcription start site (+1) and most likely acts as the position of nucleation of the RNA Pol II preinitiation complex (Figure 1⇓). The first identification of an Inr element in the MDR1 promoter came from in vitro studies, which showed that sequences from -132 to +82 were sufficient for accurate initiation, and deletion of sequences downstream of +5 decreased elongation of correctly initiated transcripts to undetectable levels (10). Subsequently, transient transfection studies indicated that sequences between -6 and +11 were sufficient for proper initiation of transcription in vivo (11, 12). Like most other “TATA-less” genes, the MDR1 promoter also includes both an inverted CCAAT element (-82 to -73) (13) and a GC-rich element (-56 to -43) that have been shown to interact with members of the Sp family of transcription factors (14–16). Transfection analyses of promoter constructs mutated in one or both of these elements indicate the involvement of the two elements in the constitutive (i.e., operative under normal growth conditions) expression of MDR1 in some cell lines. Moreover, these elements and their cognate binding factors are also involved in the activation of MDR1 transcription by a variety of inducers (see below).
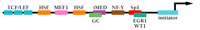
Promoter elements and DNA-binding factors involved in the transcriptional regulation of MDR1. See text for details.
A second GC-rich element has been identified (-110 to -103) that is unable to interact with Sp1 and may be the site for an unidentified protein complex (14). Interestingly, immediately downstream and overlapping this GC-rich region is an inverted MED-1 element (Multiple start site Element Downstream 1; iMED; Figure 1⇑), which was first described in the hamster MDR1 homolog, pgp1, and shown to be involved in the activation of that gene in drug-resistant cells (17). Functional disruption of iMED, either through mutation of the element or the use of a transcriptional decoy to sequester iMED-binding proteins, led to a decrease in constitutive MDR1 transcription in human neuroblastoma (17) and leukemia (18) cell lines; whether this element is also involved in induction of MDR1 transcription in resistant cells is under investigation.
Constitutive Overexpression in Drug-Resistant Cells
Cells selected for resistance to drugs involved in the MDR phenotype most often exhibit constitutive overexpression of Pgp. Although often the result of MDR1 gene amplification, such overexpression has a transcriptional component that has been verified in several laboratories. A 130-kD protein, termed MDR1 promoter–enhancing factor 1 (MEF1) has recently been shown to interact with an MDR1 promoter element to mediate upregulation. Significantly, this protein was observed in drug-resistant HL60 cells but not in the non-resistant parental cell line (19), thereby suggesting a direct role in MDR1 activation. An interesting correlate has been found in MCF-7 cells, where it appears that the same promoter element binds an inhibitory complex containing NF-κB and c-Fos in parental cells, but not in resistant MCF-7/ADR cells (20). The mechanism by which MEF1 regulates MDR1 transcription, the interplay between MEF1 and NF-κB/c-Fos, and the frequency with which these complexes are involved in MDR1 activation in drug-resistant cells remain to be determined. In other studies (21), a region of the MDR1 promoter in the drug-resistant KB/VJ300 (overexpresses Pgp at the level of transcription) and KB-C1 (overexpresses Pgp due to gene amplification) cell lines was shown to be hypomethylated relative to the promoter in parental KB3-1 cells; the mechanism underlying this intriguing correlation between methylation status and MDR1 transcript levels is not yet known.
Tumor Suppressors, Oncogenes, andMDR1 Transcription
High levels of MDR1 expression are often observed in drug-naïve tumors even when the tissue of origin exhibits little or no expression of Pgp. Hence, constitutive MDR1 gene expression may be influenced in some cells by components that are involved in malignant transformation. The altered expression of various oncogenes and tumor suppressor proteins often seen in cancer suggests that they may play a role in Pgp expression. For example, several studies have shown that wild-type p53 represses transcription from both the endogenous MDR1 gene and MDR1 reporter constructs (22–24). Our laboratory has recently shown that this repression is mediated by a direct interaction of p53 with a novel binding element within the proximal MDR1 promoter (25), and that the repressive effect may involve cell-specific cofactors. Paradoxically, several common mutant p53 proteins are able to activate, rather than repress, the MDR1 promoter (22, 26); the mechanism by which this occurs has yet to be described. It is likely that the effect of p53 on MDR1 expression in various cell lines and tumors will be influenced by the baseline expression of Pgp in the cell, the complement of endogenous p53 forms present (wild-type and/or mutant), and the levels of various modulators (e.g., protein partners, posttranslational regulators) of p53 activity.
Studies further indicate that the MDR1 gene is a target of the Ras/Raf signaling pathway (27, 28). By transfection analysis, the enhanced transcriptional response to v-Raf was mapped to the Sp1-binding site within the GC-rich element (27). Interestingly, both EGR-1, a ubiquitous immediate early factor, and WT-1, the Wilm's tumor suppressor protein, also bind within the GC-rich element at a site overlapping the binding site for Sp1; activation of MDR1 by 12-O-tetradecanoylphorbol-13-acetate (TPA) is mediated by EGR-1 (29) and suppressed by WT-1 (30). It appears, therefore, that the regulation of MDR1 expression in tumor cells by Ras/Raf and WT-1 involves a complex interplay of transcription factors within a very discrete promoter region.
Recently, a link between the tumor suppressor gene APC (adenomatous polyposis coli) and MDR1 transcription was revealed (31). Mutation of APC, which occurs as an early event in the majority of patients with hereditary and sporadic colorectal cancer, leads to the accumulation of β-catenin, which is a coactivator for the transcription complex TCF/LEF (T cell factor/lymphoid enhancer factor). TCF/LEF in turn activates the MDR1 promoter through several binding sites lying between -1813 and -275. This evidence suggests a direct correlation between mutation of APC and the aberrant overexpression of MDR1 in many colorectal cancers.
Inducible Regulation ofMDR1 Transcription
In addition to its activation during the acquisition of the multidrug resistance phenotype, the MDR1 gene is differentially expressed in normal tissues, both as a consequence of differentiation triggers and in response to environmental challenges. The highest levels of MDR1 expression are found in the adrenal gland, kidney, jejunum, colon, and the endothelial cells of the blood brain barrier and blood testes barrier (32). In addition to cell- and tissue-specific expression, environmental factors such as heat shock (33, 34), cytokines (35), differentiation agents (36-38), chemotherapeutics (39, 40), UV and x-ray irradiation (41, 42), oncogenes (22), and tumor suppressor genes (23, 24) can influence the levels of Pgp in different systems. The DNA elements and protein factors mediating the response of MDR1 to many of these stimuli have been identified.
Inducible Regulation of MDR1 in Human Tumors
Several laboratory studies have shown that Pgp expression can be transiently induced in cultured cells following exposure to chemotherapeutic agents (39, 40). We have recently shown that a similar rapid induction of Pgp occurs in patient tumors during the course of chemotherapy (43). Metastatic sarcoma tumors isolated from patients while they were undergoing chemotherapy (doxorubicin) exhibited a rapid (within ten minutes) induction of Pgp expression, with no induction observed in adjacent (lung) tissue. This response suggests a greater and heretofore unconsidered role of Pgp in the induction of, rather than the selection for, solid tumor resistance (Figure 2⇓). Moreover, the recent observations linking Pgp to a general anti-apoptotic mechanism, taken together with the transcriptional induction of MDR1 by a variety of stress inducers, indicate that Pgp may play a role in determining efficacy of therapeutic regimens (non-MDR-related) that has not previously been considered, underscoring the need for novel approaches aimed at regulating Pgp expression.
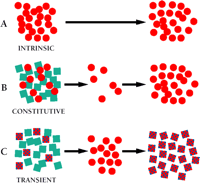
Modes of drug resistance in tumor cells. Tumor cell populations are indicated by colored geometries. Black arrows indicate the passage of time over which drug is administered.
A. Intrinsic drug resistance. The tumor is predominantly resistant to drug treatment, perhaps due to inherent properties of the tumor cells (red circles).
B. Constitutive (acquired) resistance. Within the population of tumor cells there exists, in addition to drug-sensitive cells (blue squares), a subpopulation of cells that, by virtue of mutation, have acquired an MDR phenotype (red circles). These MDR cells survive initial therapy and repopulate as a resistant tumor.
C. Transient (acquired) resistance. A subset of cells (red checked squares) within the tumor cell population are able to survive drug treatment temporarily by overexpressing the MDR1 gene. In laboratory experiments, this resistance is transient; the significance of this resistance in the clinical setting is still under investigation.
Stress Induction ofMDR1
From the earliest studies of Pgp function and regulation, MDR1 has been regarded as a stress-response gene, because its activity can be modulated by environmental adversity (44). MDR1 inducers include heat shock, the surgical insult of partial hepatectomy, exposure to carcinogens including chemotherapeutics, and UV- and x-irradiation. Although the transcriptional events that underlie these responses are not completely understood, considerable progress has been made in this area.
Heat Shock
Activation of MDR1 gene expression by heat shock and heavy metals was first reported by Gottesman and coworkers (33) and was suspected to be mediated by a single mechanism. Initially, a heat shock element (HSE) identified at position -152 to -178 was shown to be capable of binding the heat shock factor (HSF), and a second HSE at position -99 to -66 was found to bind a protein complex which was largely unchanged upon heat shock (34). However, it has recently been shown that activation by heat shock requires the interaction of HSF1 with heat shock elements within the -315 to -285 region of the MDR1 promoter, whereas activation by other stress agents, including arsenite, butyrate, and etoposide occurs independent of this HSE and HSF1 (45). Indeed, activation by other stress inducers has been shown to occur through another promoter region, which we have termed the MDR1 enhanceosome (see below).
The MDR1 Enhanceosome
It has been well established that MDR1 gene expression can be activated by UV radiation, differentiation agents such as sodium butyrate and retinoic acid, and certain chemotherapeutics (44). Recent studies in our laboratory indicate that the signals from all of these divergent stimuli converge on a region of the MDR1 promoter that we refer to as the MDR1 enhanceosome (42, 46; Z. Hu, D. Friedman, S. Thayer, S. Jin, and K.W. Scotto, unpublished work) (Figure 3⇓). This region includes binding sites for the trimeric transcription factor NF-Y and the Sp family of GC-binding transcription factors. Together, these DNA-binding proteins recruit the histone acetyltransferase PCAF to the MDR1 promoter, resulting in the acetylation of promoter-proximal histones and subsequent transcriptional activation that is likely mediated by further chromatin remodeling. Although the mechanism by which each agent transduces the signal that results in promoter activation has yet to be determined, the role of the MDR1 enhanceosome in the regulation of transcription by a variety of stimuli makes it an attractive target for therapeutic intervention.
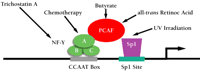
Schematic of the MDR1 enhanceosome, which includes the transcription factors NF-Y, Sp1, and PCAF.
Therapeutic Targeting ofMDR1 Transcription
To date, efforts to combat the overexpression of Pgp in the clinic have involved the use of functional modulators, or “reversal agents,” which block the Pgp-mediated efflux of anticancer agents. These modulators belong to a number of chemical classes including calcium blockers, calmodulin inhibitors, coronary vasodilators, indole alkaloids, quinolines, hormones, cyclosporines, surfactants, and antibodies (47). Although some of these agents are undergoing clinical trial, and new generations of inhibitors are being identified at a rapid pace, one of the concerns that accompanies their use is the potential interference of Pgp expression in normal cells, particularly in regard to the pharmacokinetics and biodistribution of anticancer agents. Thus, there is clear need for novel approaches to the problem of overcoming Pgp expression in tumor cells.
One promising strategy is to attack the problem of MDR1 overexpression at the level of transcription. With prophylactic intervention, one could prevent the activation of MDR1 expression, rather than block Pgp function subsequent to its overexpression. In order to supercede those results already achieved by reversal agents, the ideal drug would be one that foils transcriptional activation in tumor cells without affecting the constitutive Pgp expression required for normal physiology. Some recent studies have focused on the inhibition of MDR1 transcription as a first step toward achieving this goal.
ET-743, a Natural Chemotherapeutic that Targets MDR1 Transcription
Ecteinascidin 743 (ET-743) is a natural product isolated from the marine organism Ecteinascidia turbinata (see cover image) and is presently in phase II clinical trials for sarcoma and breast cancer in the US and Europe. ET-743 is a (tris)-tetrahydroisoquinoline, related to members of the saframycin family of compounds (Figure 4⇓) (48). Molecular modeling studies indicate that the A and B rings of ET-743 interact with the minor groove of DNA to induce a bend towards the major groove, which is a unique feature among minor groove–interacting agents (49, 50). The C subunit of ET-743 protrudes out of the helix and is potentially available for interaction with other macromolecules. Recently, we have shown that ET-743 interferes with the activation of MDR1 transcription by multiple inducers, with minimal effect on constitutive MDR1 expression even after a twenty-four-hour exposure (51).
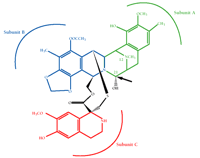
The tetrahydroisoquinoline Ecteinascidin 743 (ET-743), a novel chemotherapeutic presently in Phase II clinical trials. Subunits A and B are responsible for binding to the minor groove of DNA. The arrow points to the highly reactive carbinolamine bond responsible for alkylation. The compound is named after the marine organism, Ecteinascidia turbinata, from which it is isolated. (Figure courtesy of PharmaMar.)
Our studies indicate that ET-743 exerts its effects through the stress-responsive MDR1 enhancesome complex. Although the precise mechanism has yet to be elucidated, ET-743 does not appear to interfere with the binding of NF-Y or Sp1 to the MDR1 promoter, and thus would appear to act subsequent to complex binding and promoter-associated chromatin acetylation. Moreover, ET-743 targets the transcriptional activation of a subset of promoters that share a similar architecture to the MDR1 promoter with respect to the CCAAT and GC-rich elements (51, 52). Although this transcriptional inhibition has not yet been evaluated in animal models, our data suggest that the clinical application of ET-743 may be twofold: 1) as a novel and potent antitumor agent, and 2) as an adjuvant to first-line therapy, preventing the emergence of Pgp-induced MDR by selectively targeting transcription.
Combating MDR by Design: Therapeutic Intervention of MDR1 Transcription
Although the provocative role of ET-743 as a chemotherapeutic inhibitor of transcription has generated considerable enthusiasm in the MDR field, the ideal agent would be one that could be specifically directed to the transcription of a single gene. It is likely that the development and clinical implementation of such an agent will take several years, but initial attempts to validate this approach have been promising. Using peptide combinatorial libraries expressed in yeast, Juliano and coworkers have identified novel zinc finger proteins that bind to the overlapping EGR-1/SP1/WT-1 regulatory site in the MDR1 promoter (53). When coupled to mammalian transactivator or repressor domains, these novel proteins selectively modulate the expression of MDR1 reporter genes and inhibit the activation of the endogenous MDR1 gene. Although delivery of such peptides remains an obstacle to clinical efficacy, these studies provide the framework for a novel approach to the control of MDR1 gene–specific transcription.
Conclusions and Future Directions
The involvement of Pgp in resistance to anti-apoptotic agents, taken together with its rapid activation in response to toxic onslaught, justifies new treatment strategies aimed at preventing the aberrant upregulation of this transport protein in tumor cells. Recent advances in our understanding of the transcriptional mechanisms regulating MDR1 expression under a variety of environmental conditions have provided potential targets for novel therapeutic intervention, including the MDR1 enhanceosome, MEF1, NF-κB, Jun, and HSF. As the signaling pathways transducing toxic effects to the promoter are further revealed, additional targets will be identified. However, a number of obstacles will need to be overcome in order to bring these laboratory observations into the clinical arena. MDR1 transcriptional inhibitors need to be relatively specific, showing selectivity for tumor cells. A stable and efficient delivery system for peptide- and nucleic acid–based inhibitors must be optimized. Finally, it should be kept in mind that Pgp is only one in an ever-growing list of proteins whose aberrant expression contributes to the MDR phenotype. A better understanding of the interplay among these various transporters and apoptotic regulators, and their relative roles in the resistance exhibited by individual tumors, will be essential to the translation of these laboratory observations to the clinic.
- © American Society for Pharmacology and Experimental Theraputics 2001
REFERENCES

Kathleen W. Scotto, Ph.D., is Head of the Laboratory of Eukaryotic Transcription at the Memorial Sloan-Kettering Cancer Center and Associate Professor of Pharmcology at the Weill Graduate School of Cornell University.
Robert A. Johnson is a senior graduate student working on the transcriptional regulation of MDR1.

