
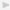

Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-α Monoclonal Antibody Trials in Systemic Lupus Erythematosus
- Yihong Yao yaoy{at}medimmune.com1
- Brandon W. Higgs higgsb{at}medimmune.com1
- Chris Morehouse morehousec{at}medimmune.com1
- Melissa de los Reyes delosreyesm{at}medimmune.com1
- Wendy Trigona trigonaw{at}medimmune.com1
- Philip Brohawn brohawnp{at}medimmune.com1
- Wendy White whitew{at}medimmune.com1
- Jianliang Zhang zhangj{at}medimmune.com1
- Barbara White whiteb{at}medimmune.com1
- Anthony J. Coyle coylea{at}medimmune.com1
- Peter A. Kiener kienerp{at}medimmune.com1
- Bahija Jallal jallalb{at}medimmune.com1
Abstract
To identify potential pharmacodynamic biomarkers to guide dose selection in clinical trials using anti-interferon-alpha (IFN-α) monoclonal antibody (mAb) therapy for systemic lupus erythematosus (SLE), we used an Affymetrix human genome array platform and identified 110 IFN-α/β-inducible transcripts significantly upregulated in whole blood (WB) of 41 SLE patients. The overexpression of these genes was confirmed prospectively in 54 additional SLE patients and allowed for the categorization of the SLE patients into groups of high, moderate, and weak overexpressers of IFN-α/β-inducible genes. This approach could potentially allow for an accurate assessment of drug target neutralization in early trials of anti-IFN-α mAb therapy for SLE. Furthermore, ex vivo stimulation of healthy donor peripheral blood mononuclear cells with SLE patient serum and subsequent neutralization with anti-IFN-α mAb or anti-IFN-α receptor mAb showed that anti-IFN-α mAb has comparable effects of neutralizing the overexpression of type I IFN-inducible genes as that of anti-IFNAR mAb. These results suggest that IFN-α, and not other members of type I IFN family in SLE patients, is mainly responsible for the induction of type I IFN-inducible genes in WB of SLE patients. Taken together, these data strengthen the view of IFN-α as a therapeutic target for SLE.
1. Introduction
The likelihood of gaining regulatory approval for new medical therapies has decreased in recent years. On average, a new drug entering phase I clinical testing is estimated to have an 8% chance of reaching the market, a decrease from the historical rate of 14% [1]. Major causes of clinical trial failures include insufficient drug activity (30%) and unacceptable toxicity profiles (30%) [2]. The development of robust pharmacodynamic (PD) markers is critical for improving the success of drugs in clinical trials and will guide selection of an optimal drug dose to balance efficacy and toxicity [2]. PD markers are often proximal in a molecular pathway to the drug target and are used to measure the effect of a drug regardless of therapeutic effect. Another important component that contributes to the success of new therapies is the development of diagnostic biomarkers that may allow better patient stratification.
Biomarkers provide more information at earlier stages of the clinical development process, thus helping to prioritize drug discovery resources and allowing for better early decisions on the fate of a development program. The US Food and Drug Administration (FDA) recently published several white papers that recognize the importance of biomarkers in drug development and clinical trials [1, 3]. While the FDA emphasized the need for biomarkers to demonstrate target neutralization, it also expressed tremendous interest in codeveloping diagnostic markers to target the correct patient population, thereby improving the drug success rate [3]. The FDA also has encouraged the adoption and integration of genomic data in drug development and regulatory assessment [4], initiating and spearheading the MicroArray Quality Consortium (MAQC) project to assess key factors contributing to the variability and reproducibility of microarray data. The MAQC has shown that microarray platforms are suitable tools to produce reliable, high-quality data that will help drug development and regulatory decision making [4–6].
Systemic lupus erythematosus (SLE) is an autoimmune disease that is characterized by severe immune system defects and the production of autoantibodies that lead to inflammation and tissue damage [7, 8]. The current standard of care involves the use of corticosteroids and toxic immunosuppressive agents that are widely acknowledged to cause unacceptable adverse events with long-term use [9]. Thus, novel therapies are needed that directly address disease pathogenesis with less toxicity. Type I interferons (IFNs) have been implicated in the development of SLE for at least 25 years [7], and elevated levels of IFN-α are detected in the serum of some SLE patients [7, 10, 11]. Previous results from microarray studies that investigated gene expression profiles in the peripheral blood of SLE patients have strengthened the idea that type I IFNs are involved in disease pathogenesis [12–14]. Furthermore, assays such as real-time polymerase chain reaction (RT-PCR) have demonstrated that overexpression of IFN-α/β-inducible genes correlated with increased disease severity and activity in SLE patients [8].
We are currently exploring an anti-IFN-α monoclonal antibody (mAb) as therapy for SLE and have used whole genome array analyses to identify putative PD and diagnostic biomarkers to aid in the development of the clinical trial. Free IFN-α protein in the serum of SLE patients would be the most reasonable choice for a PD marker for evaluating an anti-IFN-α therapy in SLE. However, our internal studies as well as others show that only a small fraction of SLE patients have measurable IFN-α protein in the sera [8, 15–17]. IFN-α-inducible genes, on the other hand, are directly downstream of the drug target, are robustly overexpressed in whole blood (WB) of the majority of SLE patients, and can be quantitatively measured by either microarray or TaqMan quantitative real-time reverse-transcriptase PCR- (QRT-PCR-) based assays [12–14].
In this study, we have used the Affymetrix human genome plus U133v2.0 array platform to examine the magnitude and prevalence of overexpression of IFN-α/β-inducible genes in WB of SLE patients. Based on these results, we selected a core group of IFN-α/β-inducible genes and confirmed the microarray results using TaqMan QRT-PCR. Furthermore, we used ex vivo stimulation of healthy donor peripheral blood mononuclear cells (PBMCs) with SLE patient serum and subsequent neutralization with anti-IFN-α mAb or anti-IFN-α receptor (IFNAR) mAb to evaluate the contribution of IFN-α to the induction of type I IFN-inducible genes in WB of SLE patients.
2. Materials and Methods
2.1. Patients and Healthy Donor Controls
Two panels of SLE patients were used in the study. The initial study panel included 41 SLE patients. WB from these SLE patients
was procured from Asterand (Detroit, Mich, USA), Cureline (South San Francisco, Calif, USA), and SeraCare (West Bridgewater,
Mass, USA). All SLE patients had a history of at least 4 of 11 positive American College of Rheumatology (ACR) classification
criteria for the diagnosis of SLE [18] and active disease manifestations at the time of sample collection. Thirty-nine (95%) were women, (mean ± SD age of 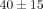 years). Thirty-two of 33 (97%) patients who were tested for the presence of anti-nuclear antibodies (ANA) came out positive.
Thirty-one of 41 (76%) SLE patients were currently receiving oral prednisone in doses ranging from 1 to 30 mg/day, with 2
SLE patients also receiving pulse intravenous steroids. More than half (24/41) of SLE patients were receiving at least 1 other
potential disease-modifying medication: hydroxychloroquine (
years). Thirty-two of 33 (97%) patients who were tested for the presence of anti-nuclear antibodies (ANA) came out positive.
Thirty-one of 41 (76%) SLE patients were currently receiving oral prednisone in doses ranging from 1 to 30 mg/day, with 2
SLE patients also receiving pulse intravenous steroids. More than half (24/41) of SLE patients were receiving at least 1 other
potential disease-modifying medication: hydroxychloroquine ( ), cyclophosphamide (
), cyclophosphamide ( ), methotrexate (
), methotrexate ( ), azathioprine (
), azathioprine ( ), cyclosporine (
), cyclosporine ( ), or mycophenolate mofetil (
), or mycophenolate mofetil ( ).
).
The prospective study panel included an independent set of SLE patient samples that was used to demonstrate a similar distribution of patients with an overexpression of IFN-α/β-inducible genes. All patients available from a phase 1a clinical trial (MI-CP126) of patients were used for the purpose. This panel included WB from 54 SLE patients from MI-CP126 investigating anti-IFN-α mAb therapy in mild-to-moderate SLE. Patients (age ≥18 years) who met at least 4 of the 11 ACR criteria for SLE were enrolled in the trial. Stable SLE background treatments with acetaminophen, nonsteroidal anti-inflammatory drugs, antimalarials, and prednisone ≤ 20 mg/day or equivalent were allowed. Patients who were receiving cyclophosphamide, azathioprine, methotrexate, mycophenolate mofetil, cyclosporine, >20 mg/day prednisone (or equivalent), immunoglobulins, blood products, investigational drugs, or antiviral therapies were excluded, as well as patients with active or chronic infection, recent vaccination with live attenuated viruses, recent herpes zoster, history of severe herpes infection, active central nervous system lupus, clinically significant cardiac, cerebrovascular, liver, or renal disease, or history of cancer. Most patients were middle-aged Caucasian females with mild to moderately active SLE with cutaneous involvement. The study was conducted according to the Declaration of Helsinki, and the study protocol was approved by the institutional review board at each site. All patients gave written informed consent before study-related procedures were performed.
The control group consisted of WB from 24 healthy normal donors (age from 23 to 56; female: male ratio is approximately 5:1) enrolled internally (MedImmune, LLC.). All the blood donors gave written informed consent for the blood to be taken and used in this study. The majority of the donors were Caucasians. Table 1 provides demographic information for the 3 groups described above.
Patient demographic information.
All WB from SLE patients and controls were collected in PAXgene RNA tubes (PreAnalytiX GmbH) according to the manufacturer's instructions.
2.2. Total RNA Extraction and Microarray Processing
Affymetrix Human Genome U133 Plus 2.0 GeneChip arrays were used in this study. Total RNA was extracted from WB samples collected in PAXgene RNA tubes using the Qiagen PAXgene Blood RNA kit (Valencia, Calif, USA). RNA purity and concentration were determined spectrophotometrically (260/280 > 1.9). The generation, fragmentation, and hybridization of biotin-labeled amplified complementary RNA (cRNA) were conducted as outlined in the Affymetrix GeneChip manual (Santa Clara, Calif, USA). The hybridizations were performed overnight and the washing/staining of arrays and scanning were carried out consistent with the standard Affymetrix protocol. Data capture and initial array quality assessment were performed with the GeneChip Operating Software.
2.3. Ex Vivo Stimulation of WB from Healthy Donors with Type I IFN Family Members
Ex vivo stimulation of WB was conducted on blood collected from 3 healthy donors enrolled internally (MedImmune, LLC.). Blood
samples (6 mL) were exposed for treatments of vehicle (1× PBS), a panel of IFN-α subtypes (IFN-α2a, −4b, −5, −6, −7, −8, −10,
−14, −16, −17), and IFN-β at concentration of 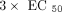 . All the cytokines were purchased from PBL Biomedical (Piscataway, NJ, USA). Following dosing, the blood was incubated at
. All the cytokines were purchased from PBL Biomedical (Piscataway, NJ, USA). Following dosing, the blood was incubated at
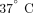 , 5%
, 5% 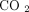 for 4 hours and transferred to a PAXgene RNA tube and inverted 8 to 10 times. The PAXgene tubes were incubated at room temperature
for 2 hours and then frozen until processed.
for 4 hours and transferred to a PAXgene RNA tube and inverted 8 to 10 times. The PAXgene tubes were incubated at room temperature
for 2 hours and then frozen until processed.
2.4. Microarray Data Analysis
ArrayAssist Lite software (Stratagene, La Jolla, Calif, USA) was used to calculate probe-level summaries (GC-RMA) from the array cell intensity files (CEL). Significance analysis of microarrays (SAMs) with control of the false discovery rate was used to select differentially regulated genes in SLE versus healthy controls using R packages (R Development Core Team, University of Auckland, New Zealand). Transcripts with a fold change ≥ 2 and q value < 0.05 were considered to be differentially regulated. Principal components analyses (PCAs) and hierarchical clustering analyses were performed using SpotFire (http://www.spotfire.com/) and R packages.
2.5. Pathway Analysis—Genego
Pathway and network analysis of gene expression data was conducted with the MetaCore integrated software suite from GeneGo, Inc. (St. Joseph, Mich, USA) using the genes determined to be significantly regulated. The significance of regulation, given a particular pathway or network, is approximated using a hypergeometric distribution in which the P value represents the probability of a particular gene set mapping arising by chance, given the (1) number of genes in the set of all genes on pathway maps, (2) genes on a particular pathway map, and (3) genes in the experiment.
2.6. TaqMan Low Density Array
The TaqMan Low Density Array (TLDA; Applied Biosystems, Foster City, Calif, USA) was used to determine the fold-change differential for a panel of 18 genes between WB of 27 SLE patients and pooled RNA from 24 healthy controls. Genes printed on the array included: 9 type I IFN-α subtypes (1, 2, 5, 6, 7, 8, 14, 17, 21), 3 additional type I IFNs (IFN-β, -β, -κ), IFN-ω, IFNαR1, IFNαR2, IFNωR1, IFNωR2, and TNF-α. Double-stranded cDNA for each patient sample was preamplified using the TaqMan PreAmp Master Mix kit (Applied Biosystems). Standard procedures for loading the array were followed and the array was run on a 7900HT Fast Real-Time PCR System (Applied Biosystems). Data analysis of the resulting Ct values was conducted with SDSv2.2.2 software (Applied Biosystems).
2.7. Fluidigm Biomark System
A mixture of 44 TaqMan Gene Expression Assays, including 4 reference control genes (Applied Biosystems), was prepared using the TaqMan PreAmp Master Mix Kit (Applied Biosystems). A total of 70 samples (35 from the 41 SLE patients in the original study and 35 from the 54 SLE patients in the prospective study) were run in triplicate (using 3 different BioMark Real-Time PCR Systems) against a set of 48 TaqMan Gene Expression Assays in BioMark 48.48 dynamic array chips (Fluidigm Corp., South San Francisco, Calif, USA). Dynamic arrays were loaded using a NanoFlex 4-IFC Controller (Fluidigm Corp.) and real-time reactions were performed using a BioMark Real-Time PCR System (Fluidigm Corp.). Results were analyzed using BioMark Real-Time PCR Analysis software. Cts above 20 were excluded from the calculation. Delta-delta Cts (ΔΔCt) were calculated using the mean of 4 reference genes (GAPDH, TFRC, β2M, and 18S) and a calibrator sample.
2.8. Ex Vivo Stimulation of PBMCs from Healthy Donors with Sera from SLE Patients
SLE serum samples were selected based on levels of type I IFN activity as determined by a reporter gene assay as previously described with some modifications [19]. Briefly, HEK293H cells were stably transfected with a luciferase construct (Gaussia princeps) under the control of the IFN-stimulated response element (ISRE). Transfected cells were incubated with 50% patient sera, and luciferase activity was detected in the culture supernatants 24 hours later. Samples generating a signal greater than 1.5 times of the negative control (normal human serum) were considered positive. To determine whether IFN-α was responsible for the positive response, cells were treated with an anti-IFN-α mAb (human IgG1; MedImmune, LLC.) and percent neutralization was calculated. Serum samples were selected for ex vivo stimulation of healthy donor PBMC based on their level of IFN-α activity.
PBMCs were harvested from WB of a healthy volunteer using Ficoll-Pacque gradient centrifugation according to manufacturer's
instructions (GE Life Sciences, Uppsala, Sweden) and were resuspended in RPMI 1640 media with GlutaMAX containing 10% fetal
bovine serum (Invitrogen, Carlsbad, Calif, USA). To measure the effects of SLE serum on the healthy donor cells, PBMCs were
cultured at a density of 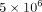 cells/mL in 250 μL/well of a 24-well plate containing 25% SLE patient serum, in the presence or absence of the following
neutralizing antibodies: anti-human-IFN-α (0.1, 1, and 10 μg/mL; human IgG1; MedImmune, LLC.), anti-human IFN-ω (10 μg/mL;
mouse IgG1, clone MMHG-1; PBL), anti-human-IFNAR1 (10 μg/mL; human IgG1; MedImmune, LLC.), and anti-HIVgp120 as a negative
control (10 μg/mL; human IgG1, MedImmune, LLC.). Following 4-hour incubation at
cells/mL in 250 μL/well of a 24-well plate containing 25% SLE patient serum, in the presence or absence of the following
neutralizing antibodies: anti-human-IFN-α (0.1, 1, and 10 μg/mL; human IgG1; MedImmune, LLC.), anti-human IFN-ω (10 μg/mL;
mouse IgG1, clone MMHG-1; PBL), anti-human-IFNAR1 (10 μg/mL; human IgG1; MedImmune, LLC.), and anti-HIVgp120 as a negative
control (10 μg/mL; human IgG1, MedImmune, LLC.). Following 4-hour incubation at 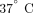 , cells were treated with Trizol LS (Invitrogen) and stored at
, cells were treated with Trizol LS (Invitrogen) and stored at 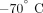 for subsequent RNA isolation.
for subsequent RNA isolation.
In a pilot study, we observed that the same SLE serum sample elicited very comparable responses in inducing the overexpression of IFN-α/β-inducible genes in PBMCs from 3 healthy donors (data not shown). This allowed us to limit the assay to 1 healthy donor PBMCs so that more SLE serum samples could be included in the study. Therefore, we selected 6 SLE serum samples based on their IFN-a bioassay results described above and used these samples to stimulate PBMCs from one healthy donor. This provided a total of 42 microarray experiments (i.e., 6 sera samples from SLE patients × 7 conditions).
3. Results
3.1. Ex Vivo Stimulation of Healthy Donor WB with IFN-α Subtypes and IFN-β
To determine the prevalence and magnitude of the overexpression of IFN-α/β-inducible genes in WB of SLE patients, we first
carried out ex vivo stimulation of healthy donor WB with different members of the type I IFN family (see Section 2) to identify IFN-α/β-inducible genes. Three samples were then subjected to transcript profiling using Affymetrix Human Genome
U133 Plus 2.0 GeneChip array. Three biological replicates of healthy donor WB were stimulated with each of the 10 IFN-α subtypes
or IFN-β (see Section 2). For each trio of cytokine treatments, a paired Student's t-test and average fold change was calculated between the three cytokine treatment replicates and the three untreated healthy
donor WB samples. Only those probes that exhibited at least a 2-fold change and 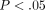 across all cytokine treatments were retained (the small sizes in each comparison restricted the use of multiple testing adjustment).
We observed that 807 and 562 transcripts were uniformly upregulated and downregulated, respectively, after stimulation of
WB of 3 healthy donors with each of 10 IFN-α subtypes or IFN-β for 4 hours.
across all cytokine treatments were retained (the small sizes in each comparison restricted the use of multiple testing adjustment).
We observed that 807 and 562 transcripts were uniformly upregulated and downregulated, respectively, after stimulation of
WB of 3 healthy donors with each of 10 IFN-α subtypes or IFN-β for 4 hours.
3.2. Overexpression of IFN-α/β-Inducible Genes Is Robust and Prevalent in WB of SLE Patients
To identify candidate PD markers for anti-IFN-α mAb clinical trials in SLE, we utilized the Affymetrix array platform to profile WB from 41 SLE patients in the initial study and 24 healthy donors. We observed that 239 and 88 transcripts were upregulated and downregulated, respectively, in WB of SLE patients compared with healthy controls. Of the 239 transcripts upregulated in WB of SLE patients, 110 were IFN-α/β-inducible (as defined by ex vivo stimulation of WB with type I IFN family members). Table 2 lists the 50 most upregulated transcripts in WB of SLE patients from the initial study; 74% of them are IFN-α/β-inducible. Table 2 also lists the prevalence of the overexpression of these genes in WB of SLE patients. These genes are overexpressed by at least 2 folds in 49% to 80% of the patients profiled. The robust and prevalent overexpression of a large number of IFN-α/β-inducible genes in SLE patients suggests that these genes might be suitable PD markers for clinical trials that investigate an anti-IFN-α mAb therapy for SLE.
Fold changes (fc; 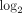 transformed) and q values (calculated using FDR) for the top 50 most upregulated transcripts in WB of SLE patients. Data were generated from
41 SLE patients in the initial study and 24 healthy controls using SAM and FDR in R (see Section 2). IFN-α/β-inducible genes are bolded. Prevalence is defined as the percentage of patients exhibiting greater than 2-fold
overexpression for a transcript compared with the baseline that is defined by the average of 24 healthy controls. FDR = false
discovery rate; IFN = interferon; SAM = significance analysis of microarrays; SLE = systemic lupus erythematosus; WB = whole
blood.
transformed) and q values (calculated using FDR) for the top 50 most upregulated transcripts in WB of SLE patients. Data were generated from
41 SLE patients in the initial study and 24 healthy controls using SAM and FDR in R (see Section 2). IFN-α/β-inducible genes are bolded. Prevalence is defined as the percentage of patients exhibiting greater than 2-fold
overexpression for a transcript compared with the baseline that is defined by the average of 24 healthy controls. FDR = false
discovery rate; IFN = interferon; SAM = significance analysis of microarrays; SLE = systemic lupus erythematosus; WB = whole
blood.
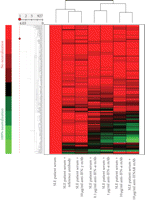
Figure 1 shows a heat map of the expression of the 110 upregulated IFN-α/β-inducible transcripts in WB of 41 SLE patients in the initial study compared to healthy controls. A total of 30/41 of the SLE patients profiled showed significant overexpression of the IFN-α/β-inducible gene signature. To quantify the magnitude of overexpression of IFN-α/β-inducible genes in WB of SLE patients, we developed an algorithm that takes advantage of the whole genome array approach. Briefly, we selected the 25 most highly overexpressed IFN-α/β-inducible genes in individual SLE patients based on the 807 IFN-α/β-inducible transcripts generated from the ex vivo stimulation of healthy donor WB study, and used the median fold change of these 25 genes to construct an IFN-α/β-inducible gene signature score for each SLE patient. Figure 2 shows the distribution of the IFN-α/β-inducible gene signature scores of the 41 SLE patients in the initial study. We classified the SLE patients into 3 groups based on their IFN-α/β-inducible gene signature score: high IFN-α/β-inducible gene signature (score > 10); moderate IFN-α/β-inducible gene signature (score 4–10); and weak IFN-α/β-inducible gene signature (score < 4). The classification of SLE patients based on IFN-α/β-inducible gene signature score is mainly for the purpose of evaluating PD in the early phases of clinical trials of anti-IFN-α mAb therapy in SLE. The SLE patients with a weak or nondetectable IFN-α/β-inducible gene signature score are unlikely to provide accurate assessment of the pharmacologic effect of anti-IFN-α mAb in these patients. Figure 3(a) shows the PCA plot of the 41 SLE patients in the initial study using the 110 overexpressed IFN-α/β-inducible transcripts. We observed a clear difference between SLE patients that had distinct overexpression of the IFN-α/β-inducible gene signature from healthy donors and SLE patients that had a weak or nondetectable IFN-α/β-inducible gene signature in WB.
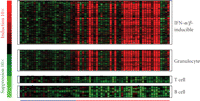
Representative heat map visualizing the overexpression of IFN-α/β-inducible gene signature, granulocyte signature, and underexpression
of T-cell and B-cell signature in WB from 41 SLE patients  compared with WB from 24 healthy donors
compared with WB from 24 healthy donors  . IFN = interferon; SLE = systemic lupus erythematosus.
. IFN = interferon; SLE = systemic lupus erythematosus.
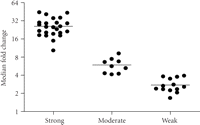
Magnitude of overexpression of IFN-α/β-inducible gene signature in WB of 41 SLE patients in the initial study as measured by the median fold change of the 25 most overexpressed IFN-α/β-inducible genes (IFN-α/β-inducible gene signature score) in individual SLE patients. The horizontal bars represent the median values. Patients whose IFN-α/β-inducible gene signature score was >10 were considered to have high IFN-α/β-inducible gene signatures; those with scores between 4 and 10 were considered to have moderate IFN-α/β-inducible gene signatures, whereas those with scores < 4 were considered to have weak IFN-α/β-inducible gene signatures. IFN = interferon; SLE = systemic lupus erythematosus.
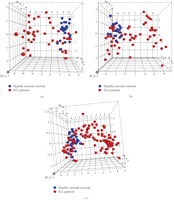
IFN-α/β-inducible genes in WB of SLE patients can be used to separate SLE patients with IFN-α/β-inducible gene signature from healthy normal controls. (a) Three-dimensional PCA plot of WB from 41 SLE patients in the initial study using the 110 upregulated IFN-α/β-inducible transcripts upregulated in WB of SLE patients compared with those from 24 healthy donors. (b) PCA plot of WB from 54 SLE patients in the prospective study using the same 110 upregulated IFN-α/β-inducible transcripts confirmed the overexpression of IFN-α/β-inducible gene signatures in SLE patients. (c) PCA plot of WB from 95 SLE samples in both discovery and prospective study using the 21 upregulated IFN-α/β-inducible gene panel in SLE patients compared with 24 healthy donors. Each point represents one sample (blue points: healthy normal controls; red points: SLE patients). IFN = interferon; PCA = principal components analysis; SLE = systemic lupus erythematosus.
To validate the observation that IFN-α/β-inducible genes are overexpressed in WB of SLE patients, we procured WB from 54 SLE patients enrolled in a prospective study. Figure 3(b) shows the PCA plot from the 54 SLE patients using the same 110 IFN-α/β-inducible transcripts identified. We observed a very similar separation of SLE patients based on the IFN-α/β-inducible gene signature as in Figure 3(a). The distribution of the IFN-α/β-inducible gene signature score in the prospective study was also similar to that of the initial study (data not shown). The ability to use the overexpressed IFN-α/β-inducible genes identified to segregate SLE patients into 2 distinct groups—patients with or without IFN-α/β-inducible gene signature—validated the accurate identification of overexpression in the IFN-α/β-inducible gene signature in WB of SLE patients.
We also observed the overexpression of a gene signature that is indicative of granulocyte activation in WB of SLE patients. This granulocyte signature was present in about 50% of the SLE patients profiled and included but was not limited to the following genes: AZU, DEFA1, DEFA4, ELA2, MMP8, MMP9, RNAS2, MPO, CAMP, FCAR, and CYBB (Figure 1). The downregulation of T and B cell gene signatures was also observed in WB of SLE patients (Figure 1), and is consistent with the observation of lymphopenia in the peripheral blood of SLE patients that has been previously reported in the literature [13, 20]. Table 3 lists the 50 most downregulated transcripts observed in WB of SLE patients.
Fold changes (fc; 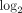 transformed) and q values (calculated using FDR) for the top 50 most downregulated transcripts in WB of SLE patients. Data were generated from
41 SLE patients in the initial study and 24 healthy controls using SAM and FDR in R (see Section 2). FDR = false discovery rate; SLE = systemic lupus erythematosus; SAM = significance analysis of microarrays; WB = whole
blood.
transformed) and q values (calculated using FDR) for the top 50 most downregulated transcripts in WB of SLE patients. Data were generated from
41 SLE patients in the initial study and 24 healthy controls using SAM and FDR in R (see Section 2). FDR = false discovery rate; SLE = systemic lupus erythematosus; SAM = significance analysis of microarrays; WB = whole
blood.
To further confirm our observation of overexpression of the IFN-α/β-inducible and granulocyte gene signatures, and to identify other signaling pathways that may be altered in SLE, we carried out a pathway and network analysis with GeneGo software (see Section 2). Overall, this pathway analysis confirmed the activation of the type I IFN signaling pathway, along with the activation of granulocytes and the downregulation of T-cell signaling pathways in SLE. The interleukin (IL)-10 signaling pathway was among other notable pathways found to be activated or, otherwise, altered in the SLE patients who were profiled. This is likely due to the abnormal apoptosis of T-cell subsets observed in SLE patients [21, 22].
3.3. Confirmation of the Overexpression of IFN-α/β-Inducible Genes Identified by Microarrays Using TaqMan QRT-PCR Assays
To confirm the overexpression of IFN-α/β-inducible genes in WB of SLE patients, which was observed in the microarray analyses,
we used a BioMark 48.48 dynamic array to perform the high-throughput TaqMan QRT-PCR on the top 40 most overexpressed IFN-α/β-inducible
genes in WB of SLE patients. The overexpression of all these genes was confirmed by TaqMan QRT-PCR assays in WB of 35 of the
41 SLE patients randomly selected from the original study, and also 35 of the 54 SLE patients selected from the prospective
study. The majority of the data showed a strong correlation between microarray and TaqMan assays. The overexpression of 15
of the 40 IFN-α/β-inducible genes using TaqMan assays is shown in Figure 4(a). These genes were upregulated by an average of 8- to 92-fold, and all were significantly overexpressed (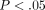 ).
).
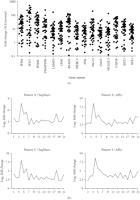
TaqMan QRT-PCR confirmed the overexpression of IFN-α/β-inducible genes in WB of SLE patients. (a) Relative fold changes of
15 IFN-α/β-inducible genes (out of the 40 assayed) in SLE patients were compared with healthy donors (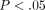 for all). Averages of relative mRNA levels of genes in the pooled RNA from 24 healthy donors were scaled to 1 based on TaqMan
QRT-PCR assays. Horizontal bars represent average fold change. (b) TaqMan QRT-PCR validation of overexpression of the 21-gene
panel of IFN-α/β-inducible genes in WB of SLE patients as determined by whole genome array. The relative overexpression of
21 IFN-α/β-inducible genes in 2 SLE patients is shown via (left) microarray and (right) TaqMan assays. Correlation coefficients
(r) between TaqMan QRT-PCR and microarray were 0.986 and 0.989 for patient X and Y, respectively. IFN = interferon; QRT-PCR
= quantitative real-time reverse transcriptase polymerase chain reaction; SLE = systemic lupus erythematosus.
for all). Averages of relative mRNA levels of genes in the pooled RNA from 24 healthy donors were scaled to 1 based on TaqMan
QRT-PCR assays. Horizontal bars represent average fold change. (b) TaqMan QRT-PCR validation of overexpression of the 21-gene
panel of IFN-α/β-inducible genes in WB of SLE patients as determined by whole genome array. The relative overexpression of
21 IFN-α/β-inducible genes in 2 SLE patients is shown via (left) microarray and (right) TaqMan assays. Correlation coefficients
(r) between TaqMan QRT-PCR and microarray were 0.986 and 0.989 for patient X and Y, respectively. IFN = interferon; QRT-PCR
= quantitative real-time reverse transcriptase polymerase chain reaction; SLE = systemic lupus erythematosus.
3.4. mRNAs of Type I IFN Family Members are Overexpressed in SLE Patients Using TaqMan QRT-PCR Assays
Given that we observed significant overexpression of IFN-α/β-inducible genes in WB of SLE patients, we wanted to characterize
the type I IFNs that may be responsible for this upregulation. Since the type I IFN protein can only be measured in a small
fraction of SLE patients, we used the TLDA technology from Applied Biosystems to measure the mRNA level of type I IFN family
members in WB of 27 SLE patients, and compared that with pooled RNA from WB of 24 healthy donors. We found that the overexpression
of mRNAs of 9 IFN-α subtypes in WB of SLE patients was significant (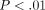 ) compared with healthy controls (Figure 5(a)). In addition, the mRNAs of other type I IFN family members, such as IFN-β and IFN-κ, were also significantly overexpressed
in SLE (
) compared with healthy controls (Figure 5(a)). In addition, the mRNAs of other type I IFN family members, such as IFN-β and IFN-κ, were also significantly overexpressed
in SLE (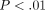 ), as were the type I IFN receptors IFNAR1 and IFNAR2 (Figure 5(b)). These observations suggest that upregulation of mRNAs of type I IFN family members may contribute to the overexpression
of their respective proteins, which may in turn underscore the overexpression of IFN-α/β-inducible gene signature in WB of
SLE patients. Furthermore, we observed that TNF-α, IFN-ω, IFNGR1, and IFNGR2 transcripts were also upregulated in WB of SLE
patients (Figure 5(c)). However, the relative magnitude of overexpression of these transcripts was much less than those of type I IFN family
members, especially the IFN-α subtypes (Figures 5(a) and 5(b)).
), as were the type I IFN receptors IFNAR1 and IFNAR2 (Figure 5(b)). These observations suggest that upregulation of mRNAs of type I IFN family members may contribute to the overexpression
of their respective proteins, which may in turn underscore the overexpression of IFN-α/β-inducible gene signature in WB of
SLE patients. Furthermore, we observed that TNF-α, IFN-ω, IFNGR1, and IFNGR2 transcripts were also upregulated in WB of SLE
patients (Figure 5(c)). However, the relative magnitude of overexpression of these transcripts was much less than those of type I IFN family
members, especially the IFN-α subtypes (Figures 5(a) and 5(b)).
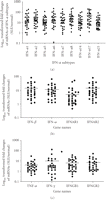
Relative expression of mRNAs and median fold changes (horizontal bars) of (a) type I IFN-α subtypes, (b) other members of
the type I IFNs and IFN-α receptors, and (c) TNF-α, IFN-ω, and IFN-ω receptors in WB of SLE patients compared with healthy
controls (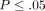 for all). Averages of relative mRNA levels of these cytokines and their receptors in WB from 24 healthy donors were scaled
to 1 based on TaqMan QRT-PCR assays. IFN = interferon; QRT-PCR = quantitative real-time reverse transcriptase polymerase chain
reaction; SLE = systemic lupus erythematosus; TNF-α = tumor necrosis factor.
for all). Averages of relative mRNA levels of these cytokines and their receptors in WB from 24 healthy donors were scaled
to 1 based on TaqMan QRT-PCR assays. IFN = interferon; QRT-PCR = quantitative real-time reverse transcriptase polymerase chain
reaction; SLE = systemic lupus erythematosus; TNF-α = tumor necrosis factor.
3.5. Identification of a Panel of IFN-α/β-Inducible Genes That are Neutralized by an Anti-IFN-α mAb
Among the 807 IFN-α/β-inducible transcripts originally identified, we aimed to eliminate genes that were likely to be upregulated by multiple cytokines in SLE or poorly neutralized by anti-IFN-α mAb in SLe (rendering them poor candidates as PD markers in clinical trials investigating anti-IFN-α mAb therapy in SLE). One of the approaches to address these issues was to identify the IFN-α/β-inducible genes induced in healthy donor PBMC ex vivo by SLE patient sera that were also neutralized by anti-IFN-α mAb.
Overall, sera from 6 SLE patients were characterized based on their level of IFN-α activity as measured in an ISRE reporter
gene assay, and were used to stimulate PBMC of one healthy donor ex vivo. There was a positive correlation between the IFN-α
activity in serum of SLE patients and the magnitude of IFN-α/β-inducible genes induced as measured by the IFN-α/β-inducible
gene signature score (data not shown). A total of 436 of 807 IFN-α/β-inducible transcripts were upregulated by more than 2
folds when challenged with at least one SLE patient serum. Of these 436 transcripts, the overexpression of 161 was inhibited
≥50% when treated with the highest dose of anti-IFN-α mAb (6 total samples), and inhibition of ≥70% for any sample treated
with the single dose of anti-IFNAR1 mAb (6 total samples). The heat map demonstrating the effects of anti-IFN-α and -ω and
anti-IFNAR1 mAbs on the genes upregulated in healthy donor PBMC by treatment with the serum of one SLE patient is shown in
Figure 6. The anti-IFN-α mAb treatment (lanes 4–6) demonstrated a strong neutralizing effect on a large number of genes stimulated
with the serum of an SLE patient. Furthermore, the neutralizing effect of the anti-IFN-α mAb was dose-dependent, as evaluated
by the differences in the number of transcripts that were inhibited by treatment with anti-IFN-α mAb ≥50% at each of the 3
dosage levels (0.1, 1, and 10 μg/mL) within each of the 6 SLE patient serum samples. For example, the mean ± SD normalized
ratios of the number of inhibited transcripts ≥50% between 0.1, 1, and 10 μg/mL treatments of anti-IFN-α mAb and 10 μg/mL
treatment of anti-IFNAR mAb are 1.00, 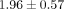 ,
, 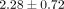 , and
, and 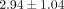 , respectively. This suggests that these genes might be good candidates for PD markers for clinical trials evaluating anti-IFN-α
mAb therapy in SLE. The control mAb inhibited the overexpression of some genes upregulated when challenged with SLE patient
sera (including IFN-α/β-inducible genes) (lane 2). However, the effect of the anti-IFN-α mAb was much broader, with a strong
neutralizing effect observed in a large number of genes in which neither the reference mAb nor anti-IFN-ω mAb had any significant
effect (lanes 2-3; lanes 4–6). When examining the mean ± SD percentage of genes inhibited ≥50% by each of the antibody treatments,
there is a much stronger effect on gene counts for the 10 μg/mL treatments of anti-IFN-α mAb and anti-IFNAR mAb (
, respectively. This suggests that these genes might be good candidates for PD markers for clinical trials evaluating anti-IFN-α
mAb therapy in SLE. The control mAb inhibited the overexpression of some genes upregulated when challenged with SLE patient
sera (including IFN-α/β-inducible genes) (lane 2). However, the effect of the anti-IFN-α mAb was much broader, with a strong
neutralizing effect observed in a large number of genes in which neither the reference mAb nor anti-IFN-ω mAb had any significant
effect (lanes 2-3; lanes 4–6). When examining the mean ± SD percentage of genes inhibited ≥50% by each of the antibody treatments,
there is a much stronger effect on gene counts for the 10 μg/mL treatments of anti-IFN-α mAb and anti-IFNAR mAb (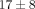 % and
% and 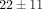 % genes inhibited, resp.), as compared to the treatments of anti-IFN-ω and the control mAb (
% genes inhibited, resp.), as compared to the treatments of anti-IFN-ω and the control mAb (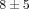 % and
% and 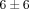 % genes inhibited, resp.). It should be noted that treatment with anti-IFNAR1 mAb (lane 7) induced a greater neutralization
than anti-IFN-α mAb, suggesting the possible presence (although of minor effect) of other type I IFN family members in addition
to IFN-α in the serum of the SLE patient.
% genes inhibited, resp.). It should be noted that treatment with anti-IFNAR1 mAb (lane 7) induced a greater neutralization
than anti-IFN-α mAb, suggesting the possible presence (although of minor effect) of other type I IFN family members in addition
to IFN-α in the serum of the SLE patient.
Representative heat map demonstrating anti-IFN-α, -IFNAR, and -IFN-ω mAb effects on healthy donor PBMC stimulated with serum from 1 SLE patient. Lane 1: SLE patient serum only; Lane 2: SLE patient serum plus reference antibody; Lane 3: SLE patient serum plus 10 μg/mL anti-IFN-ω mAb; Lanes 4–6: SLE patient serum plus increasing concentrations of anti-IFN-α mAb (0.1, 1, and 10 μg/mL); Lane 7: SLE patient serum plus 10 μg/mL anti-IFNAR mAb. Color represents relative neutralization (inhibition) of overexpression of individual genes upregulated by soluble mediators in the serum of an SLE patient. The red color represents no neutralization, and green represents neutralization of overexpression of individual genes. IFN = interferon; IFNAR = interferon associated receptor; PBMC = peripheral blood mononuclear cells; SLE = systemic lupus erythematosus.
3.6. Selection of a 21-Gene Panel of IFN-α/β-Inducible Genes as Potential PD and Diagnostic Biomarkers to Validate in Clinical Trials
To select a small, robust panel of IFN-α/β-inducible genes that could be developed into a high-throughput PD marker assay
to measure anti-IFN-α mAb effect in SLE, we narrowed the gene panel to 21 genes so that they could be assayed by either TLDAs
or Fluidigm BioMark 48.48 dynamic array chips. The process for the selection of 21 IFN-α/β-inducible genes as candidate PD
markers to measure anti-IFN-α mAb therapy in SLE is outlined in Figure 7. Briefly, we started with 807 IFN-α/β-inducible transcripts identified byex vivo stimulation of WB of 3 healthy donors with
10 IFN-α subtypes and IFN-β. Then, we identified that 110 overexpressed transcripts (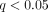 ; fold change ≥ 2) in WB of 41 SLE patients in the initial study were IFN-α/β-inducible using SAM and FDR.
; fold change ≥ 2) in WB of 41 SLE patients in the initial study were IFN-α/β-inducible using SAM and FDR.
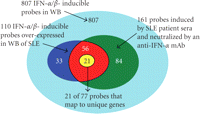
Venn diagram illustrating the three primary analyses used in the selection process of 21 candidate PD markers for anti-IFN-α mAb therapy in SLE: (1) 807 IFN-α/β-inducible transcripts determined from ex vivo stimulation of healthy donor WB with 10 IFN-α subtypes and IFN-β (cyan region); (2) 110 transcripts found to be both overexpressed in WB of SLE patients and IFN-α/β-inducible in WB of healthy donors (combination of blue, yellow, and red regions); (3) 161 transcripts identified by ex vivo stimulation to be induced by SLE patient sera and subsequently neutralized by an anti-IFN-α mAb (combination of green, yellow, and red regions). The intersection of these three analyses provided a list of 77 transcripts, which were ranked by magnitude and prevalence across SLE patients (i.e., percentage of SLE patients with a fold change of at least 2) and the top 21 unique genes were chosen. IFN = interferon; SLE = systemic lupus erythematosus.
To identify whether these genes could be neutralized by an anti-IFN-α mAb in SLE, we stimulated 1 healthy donor PBMC ex vivo with sera from 6 individual SLE patients. We observed that 161 (of the 807 transcripts) IFN-α/β-inducible transcripts were upregulated by ≥2 folds in the PBMC of the healthy donor following stimulation with at least 1 SLE patient serum in which the overexpression of these genes was suppressed by ≥50% and ≥70% by an anti-IFN-α mAb and an anti-IFN-αR mAb, respectively.
77 transcripts were common to this list of 161 transcripts (identified in the neutralization experiments) and the previously
determined list of 110 transcripts (identified to be overexpressed in WB of 41 SLE patients). These transcripts are both IFN-α/β-inducible
and can be neutralized by an anti-IFN-α mAb. Each of the 77 transcripts was ranked by the average fold-change magnitude across
all SLE patients and the percentage of patients displaying a change ≥2 folds. The 21 most prevalently overexpressed IFN-α/β-inducible
genes (that represent unique genes using the NetAffx annotation file for the Affymetrix U133v2.0 plus array; ESTs were excluded)
from this ranking were selected as candidate PD markers for anti-IFN-α therapy in SLE. Four genes: OAS2, MX1, PLSCR1, and
DNAPTP6 were chosen over a few other candidate genes that showed slightly higher overexpression in WB of SLE patients due
to the strong indication from the literature of their involvement in SLE, antiviral response, or involvement in type I IFN
signaling pathway [23–25]. A 21-gene panel was chosen so that the high-throughput TaqMan assays can be carried out on the TDLA array. Table 4 lists the 21 IFN-α/β-inducible genes in WB of 95 SLE patients from both initial and prospective studies described earlier.
The consistency of the results in both microarray and TaqMan assays and the strong correlation (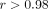 ) between microarray and TaqMan assays for 21 IFN-α/β-inducible genes in the 2 example SLE patients (Figures 4(b)) provide more evidence that these genes may be useful as PD and diagnostic markers of anti-IFN-α treatment in SLE as they
are robustly measured using multiple assay platforms.
) between microarray and TaqMan assays for 21 IFN-α/β-inducible genes in the 2 example SLE patients (Figures 4(b)) provide more evidence that these genes may be useful as PD and diagnostic markers of anti-IFN-α treatment in SLE as they
are robustly measured using multiple assay platforms.
Fold changes (fc; 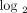 transformed) and q values (calculated using FDR) for the 21 candidate PD markers in WB of 95 SLE patients analyzed in the study. Data were generated
from 95 SLE patients from both the initial study and the prospective study and 24 healthy controls using SAM and FDR in R
(see Section 2). FDR = false discovery rate; SLE = systemic lupus erythematosus; SAM = significance analysis of microarrays; WB = whole
blood.
transformed) and q values (calculated using FDR) for the 21 candidate PD markers in WB of 95 SLE patients analyzed in the study. Data were generated
from 95 SLE patients from both the initial study and the prospective study and 24 healthy controls using SAM and FDR in R
(see Section 2). FDR = false discovery rate; SLE = systemic lupus erythematosus; SAM = significance analysis of microarrays; WB = whole
blood.
With these 21 genes, it was necessary to recalculate the thresholds of IFN-α/β-inducible gene signature score in WB of SLE
patients that were previously identified for partitioning SLE patients into high, moderate, or weak IFN-α/β-inducible gene
signatures (based on the top 25 overexpressed IFN-α/β-inducible genes in WB of individual SLE patients as measured by the
Affymetrix whole genome array) for a lower density, but high-throughput platform (TaqMan-based assay). A scaling method was
needed to convert the IFN-α/β-inducible gene signature score based on the top 25 most overexpressed IFN-α/β-inducible genes
of each SLE patient on the Affymetrix platform to the IFN-α/β-inducible gene signature score based on 21 genes selected for
all SLE patients for the TaqMan-based assay. This method was implemented to compensate for 3 primary differences between the
2 platforms: (1) the number of transcripts used for the IFN-α/β-inducible gene signature (25 genes dynamically determined
for each patient on the Affymetrix platform versus a static 21-gene list on the TaqMan-based assay), (2) the differences in
sensitivity between the 2 platforms, and (3) the scales of the dynamic ranges within each platform. First, fold-change values
were calculated (on a 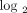 scale) for the 807 IFN-α/β-inducible transcripts between 35 SLE patients (randomly selected from the 41 SLE patients whose
microarray results were confirmed by TaqMan QRT-PCR; TaqMan QRT-PCR was also used to confirm microarray results for 35 SLE
patients chosen from the 54 SLE patients in the prospective study), and the average of a set of normal healthy controls. The
top 25 most upregulated genes based on fold-change values were determined for each patient on the Affymetrix platform (this
gene set is allowed to vary from patient to patient depending on which IFN-α/β-inducible genes are most overexpressed). Next,
the median fold change was calculated from the top 25 genes for each SLE patient. The same calculation was conducted across
identical patients using the static 21 gene set on the TaqMan-based assay. This gene set was identical for each patient, and
the median fold change was calculated. A simple regression model was then computed using these 2 vectors of equal length (35
median fold-change values), and the coefficients from the model were used to determine the conversion factor (from the Affymetrix
platform to the TaqMan-based assay;
scale) for the 807 IFN-α/β-inducible transcripts between 35 SLE patients (randomly selected from the 41 SLE patients whose
microarray results were confirmed by TaqMan QRT-PCR; TaqMan QRT-PCR was also used to confirm microarray results for 35 SLE
patients chosen from the 54 SLE patients in the prospective study), and the average of a set of normal healthy controls. The
top 25 most upregulated genes based on fold-change values were determined for each patient on the Affymetrix platform (this
gene set is allowed to vary from patient to patient depending on which IFN-α/β-inducible genes are most overexpressed). Next,
the median fold change was calculated from the top 25 genes for each SLE patient. The same calculation was conducted across
identical patients using the static 21 gene set on the TaqMan-based assay. This gene set was identical for each patient, and
the median fold change was calculated. A simple regression model was then computed using these 2 vectors of equal length (35
median fold-change values), and the coefficients from the model were used to determine the conversion factor (from the Affymetrix
platform to the TaqMan-based assay; 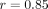 ) for the response threshold values to partition the SLE patients into an IFN-α/β-inducible gene signature of high (>10 on
Affymetrix; >5.53 on TaqMan), moderate (between 4 and 10 on Affymetrix; between 1.91 and 5.53 on TaqMan), or weak (<4 on Affymetrix;
<1.91 on TaqMan). Using these scaled threshold values, the categorized signature levels (high, moderate, or weak) that were
determined using the 21 genes from the TaqMan-based assay were comparable to those that were determined based on the top 25
upregulated IFN-α/β-inducible genes (although it should be noted that the threshold values between the 2 platforms are presented
on different scales).
) for the response threshold values to partition the SLE patients into an IFN-α/β-inducible gene signature of high (>10 on
Affymetrix; >5.53 on TaqMan), moderate (between 4 and 10 on Affymetrix; between 1.91 and 5.53 on TaqMan), or weak (<4 on Affymetrix;
<1.91 on TaqMan). Using these scaled threshold values, the categorized signature levels (high, moderate, or weak) that were
determined using the 21 genes from the TaqMan-based assay were comparable to those that were determined based on the top 25
upregulated IFN-α/β-inducible genes (although it should be noted that the threshold values between the 2 platforms are presented
on different scales).
Figure 8 shows the stratification of 35 SLE patients in the initial study into groups of expressing high, moderate, and weak IFN-α/β-inducible
gene signatures in WB based on the distribution of fold-change values (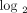 scale) of all 21 IFN-α/β-inducible genes. The median fold change of the 21 genes for each patient (as measured by the dynamic
array from Fluidigm) was used to partition each patient into these 3 groups. The vertical dashed lines partitioned the 3 classes
of IFN-α/β-inducible gene signature scores: 7 patients with a weak IFN-α/β-inducible gene signature = median fold change <
1.91 (0.93 on
scale) of all 21 IFN-α/β-inducible genes. The median fold change of the 21 genes for each patient (as measured by the dynamic
array from Fluidigm) was used to partition each patient into these 3 groups. The vertical dashed lines partitioned the 3 classes
of IFN-α/β-inducible gene signature scores: 7 patients with a weak IFN-α/β-inducible gene signature = median fold change <
1.91 (0.93 on 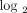 scale), 8 patients with a moderate IFN-α/β-inducible gene signature = median fold change between 1.91 and 5.53, and 20 patients
with a strong IFN-α/β-inducible gene signature = median fold change > 5.53 (2.47 on
scale), 8 patients with a moderate IFN-α/β-inducible gene signature = median fold change between 1.91 and 5.53, and 20 patients
with a strong IFN-α/β-inducible gene signature = median fold change > 5.53 (2.47 on 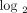 scale). In a PCA plot for all SLE patients profiled in this study (
scale). In a PCA plot for all SLE patients profiled in this study ( ) and for the 24 healthy control samples using the 21 IFN-α/β-inducible genes, a clear distinction between SLE patients with
an overexpressed IFN-α/β-inducible gene signature and those with weak or nondetectable IFN-α/β-inducible gene signatures was
observed (Figure 3(c)). Furthermore, the SLE patients with weak or nondetectable IFN-α/β-inducible gene signatures were found to cluster with
healthy donors. Importantly, the partitioning between these groups using the 21-gene panel of IFN-α/β-inducible genes was
similar to that observed using the larger 110-gene set (Figures 3(a) and 3(b)).
) and for the 24 healthy control samples using the 21 IFN-α/β-inducible genes, a clear distinction between SLE patients with
an overexpressed IFN-α/β-inducible gene signature and those with weak or nondetectable IFN-α/β-inducible gene signatures was
observed (Figure 3(c)). Furthermore, the SLE patients with weak or nondetectable IFN-α/β-inducible gene signatures were found to cluster with
healthy donors. Importantly, the partitioning between these groups using the 21-gene panel of IFN-α/β-inducible genes was
similar to that observed using the larger 110-gene set (Figures 3(a) and 3(b)).
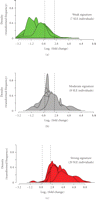
Stratification of 35 SLE patients into groups expressing low ((a) green), moderate ((b) gray), and high ((c) red) IFN-α/β-inducible
gene signaturebased onmedian fold change across the 21-gene panel of IFN-α/β-inducible genes. Kernel density estimates (i.e.,
histograms or frequency plots) for each SLE individual are calculated and graphed using the fold change foreach of the 21
genes from each SLE patient on the 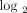 scale to provide a representation of the distribution of 21 gene fold change values. The vertical dashed lines partition
the 3 classes of IFN-α/β-inducible gene signature scores: 7 individuals with a weak IFN-α/β-inducible gene signature = median
fold change <1.91 (0.93 on
scale to provide a representation of the distribution of 21 gene fold change values. The vertical dashed lines partition
the 3 classes of IFN-α/β-inducible gene signature scores: 7 individuals with a weak IFN-α/β-inducible gene signature = median
fold change <1.91 (0.93 on 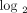 scale); 8 individuals with a moderate IFN-α/β-inducible gene signature = median fold change between 1.91 and 5.53; and 20
individuals with a strong IFN-α/β-inducible gene signature = median fold change >5.53 (2.47 on
scale); 8 individuals with a moderate IFN-α/β-inducible gene signature = median fold change between 1.91 and 5.53; and 20
individuals with a strong IFN-α/β-inducible gene signature = median fold change >5.53 (2.47 on 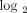 scale). IFN = interferon; SLE = systemic lupus erythematosus.
scale). IFN = interferon; SLE = systemic lupus erythematosus.
We also assessed the difference in variability between the 24 normal healthy controls and the SLE patients for the 21 IFN-α/β-inducible genes selected; we conducted a variance assessment using the three categorized IFN-α/β-inducible gene signature levels. We compared the variance of each gene between the normal healthy controls and two groups of SLE patients: SLE patients with a weak IFN-α/β-inducible gene signature score (values < 4) and all SLE patients. The reasoning for comparing the normal healthy controls to those SLE patients with a weak IFN-α/β-inducible gene signature score was to evaluate the normal control variability against a set of patients that have comparable magnitude of IFN-α/β-inducible gene signature score. We would expect the variance for each of the 21 genes within the normal controls and the SLE patients with a weak IFN-α/β-inducible gene signature score to be similar.
We used an F-test to assess differences in variance for each of the 21 genes individually between the 2 groups. Using a Bonferroni-adjusted
threshold of 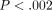 (0.05/21), a total of 2/21 genes with significant differences in variance between normal healthy controls and SLE patients
with a weak IFN-α/β-inducible gene signature score were observed, and 6/21 genes with a significant difference in variance
between normal healthy controls and all SLE patients were identified. All genes with significant differences in variance had
a lower variance in the normal healthy control group. This analysis suggests that the variability is lower among the 24 normal
healthy control samples when compared to the SLE patient samples for these 21 genes.
(0.05/21), a total of 2/21 genes with significant differences in variance between normal healthy controls and SLE patients
with a weak IFN-α/β-inducible gene signature score were observed, and 6/21 genes with a significant difference in variance
between normal healthy controls and all SLE patients were identified. All genes with significant differences in variance had
a lower variance in the normal healthy control group. This analysis suggests that the variability is lower among the 24 normal
healthy control samples when compared to the SLE patient samples for these 21 genes.
4. Discussion
The identification of biomarkers that can assist in the execution and interpretation of clinical trials may involve the detection of unique molecular signatures that correlate with biological events [26]. In developing drugs against cytokines and chemokines where it is difficult to measure the protein in the serum of patients, it is necessary to use biomolecules (proteins and transcripts) that are directly downstream of the drug targets to measure the pharmacologic effect of these drugs when they can be accurately measured. In this study, we have used a 3-tiered approach to identify potential PD and diagnostic markers for clinical trials investigating anti-IFN-α mAb treatments in SLE. The first tier involved characterizing the biological variation (patient-to-patient variation) among SLE patients by identifying genomic biomarkers with whole genome microarray analyses from a training panel of SLE patients and then confirming the validity of those markers in a separate, prospective panel of SLE patients. Because of the expensive nature of microarray analyses, the development of an assay that could be performed on a high-throughput platform was of utmost importance, so it could be used in later phases of clinical trials in which several thousand samples may be routinely assayed. Therefore, the second tier involved validating the findings from microarray analyses in which TaqMan-based assays were performed and optimized for use in a premier high-throughput platform (BioMark 48.48 dynamic array from Fluidigm). In all the assays performed to date, the platform provided sensitive and robust results, with intra-array variations of ≤2% and interarray variations of ≤5%. To further enhance the specificity of the assay, the third tier of our approach involved the narrowing of the number of genes to be analyzed from 807 to 77 and, finally, to 21 IFN-α/β-inducible genes that were consistently and markedly overexpressed in WB of SLE patients. This reduction in the number of genes in the screening process allowed for the simplification of the analysis of the results and the increase of throughput. The robust and prevalent overexpression of these 21 IFN-α/β-inducible genes in SLE patients, coupled with the fact that these genes are directly downstream of type I IFN, suggest that they may be well suited as PD markers in clinical trials targeting IFN-α. Additionally, the ability to use these genes to differentiate between SLE patients with moderate-to-high overexpression of IFN-α/β-inducible gene signature from those with weak or nondetectable signature and healthy normal controls suggests the use of these genes as possible diagnostic markers in clinical trials. This may be especially true if the clinical benefits of anti-IFN-α mAb therapy occur primarily in SLE patients who significantly overexpress the IFN-α/β-inducible genes in WB (Figure 3(c)). This hypothesis needs to be validated in the clinical trials. To capture the magnitude of IFN-α/β effects in WB of SLE patients, we developed an algorithm to calculate the IFN-α/β-inducible gene signature scores using either a static 21 gene or a dynamic top 25 gene list in WB of SLE patients. Results from these two methods agree with each other very well (correlation coefficient of 0.96), and also agree with the IFN scores as described by Feng et al. [23] (correlation coefficients are 0.95 and 0.92 between Feng's method and a static 21 gene or a dynamic 25 gene algorithm, resp., for calculating an IFN score).
SLE is an autoimmune disease characterized by the involvement of many different organ systems and by immunologic abnormalities, such as the accumulation of autoantibodies. Type I IFNs have been implicated in the pathogenesis of SLE and some patients periodically demonstrate elevated serum levels of type I IFNs. Furthermore, clinical observations have suggested a role for type I IFNs in the development of SLE; SLE symptoms have presented in patients with cancer or viral infections who received recombinant IFN-α therapy [27]. In recent years, microarray analyses have provided evidence for the measurement of type I IFN in SLE that the enhanced expression of a number of IFN-α/β-inducible genes has been observed in the peripheral blood of SLE patients [12, 13, 28]. The study described in this paper is the largest study to date that has evaluated the type I IFN effect in the periphery of SLE using a genomics approach. The ex vivo stimulation of healthy donor PBMC with SLE patient serum samples and subsequent neutralization with anti-IFN-α mAb or anti-IFNAR mAb show that anti-IFN-α mAb has comparable effects of neutralizing the overexpression of type I IFN-inducible genes as that of anti-IFNAR mAb. These results suggest that it is IFN-α, not other members of type I IFN family in the serum of SLE patients, which is mainly responsible for the induction of type I IFN-inducible genes in WB of SLE patients.
SLE patients in this study were able to be classified as expressing high, moderate, or weak IFN-α/β-inducible gene signatures in the periphery. This approach will allow us to obtain a more accurate readout on drug target neutralization in early phases of clinical trials of anti-IFN-α mAb therapy in SLE (patients with high-and-moderate overexpression of IFN-α/β-inducible gene signatures are likely to provide a more accurate assessment on PD) so that an optimal dosing regimen can be identified for use in pivotal trial. Recently, Anderson suggested a road map for the creation of a viable diagnostic marker, which is composed of the following steps: discovery, verification/validation, and clinical implementation [29]. Currently, we are evaluating the utility of these IFN-α/β-inducible genes as potential diagnostic markers to identify SLE patients that might respond to anti-IFN-α mAb therapy in several ongoing trials.
In summary, the findings described in this study provide strong scientific evidence of IFN-α as a therapeutic target in SLE. We also feel that the overexpression of IFN-α/β-inducible genes, if rigorously quantified and validated, may form the basis for developing PD and diagnostic markers in different stages of clinical trials of anti-IFN-α mAb therapy in SLE. Overall, these analyses are likely to provide valuable information during the drug development process to assist in understanding the disease mechanism and in the selection of the most appropriate patient population to achieve rapid and predictable outcomes.
Acknowledgments
The authors would like to thank Rhonda Croxton and Lauren Gallagher for editorial assistance with this manuscript, Jonathan Zmuda and Jonathan Hirsch for providing technical assistance, Chris Heid, Martin Pieprzyk, Mike Lucero, and John Lynch from Fluidigm Corporation for their assistance with large-scale TaqMan QRT-PCR assays, and Eric Phan, Krystal Bowers, and Denise Dawson for sample management. All authors are employees of MedImmune, LLC. The work described in this article was supported by MedImmune, LLC.
- Received June 19, 2008.
- Revision received October 20, 2008.
- Accepted November 5, 2008.
- © 2009 Yihong Yao et al.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- Díaz-Alderete A.,
- Crispin J. C.,
- Vargas-Rojas M. I.,
- Alcocer-Varela J., jalcocer{at}quetzal.innsz.mx
 cells in patients with systemic lupus erythematosus”. Journal of Autoimmunity, vol. 23 no. 4 pp. 379–383 2004 doi:10.1016/j.jaut.2004.10.001.
cells in patients with systemic lupus erythematosus”. Journal of Autoimmunity, vol. 23 no. 4 pp. 379–383 2004 doi:10.1016/j.jaut.2004.10.001. - ↵
- ↵
-
- Ye S., yeshuang{at}online.sh.cn
- Guo Q.,
- Tang J.-P.,
- Yang C.-D.,
- Shen N.,
- Chen S.-L.
 -oligoadenylate synthetase isoforms be biomarkers to differentiate between disease flare and infection in lupus patients?
A pilot study”. Clinical Rheumatology, vol. 26 no. 2 pp. 186–190 2007 doi:10.1007/s10067-006-0260-z.
-oligoadenylate synthetase isoforms be biomarkers to differentiate between disease flare and infection in lupus patients?
A pilot study”. Clinical Rheumatology, vol. 26 no. 2 pp. 186–190 2007 doi:10.1007/s10067-006-0260-z. - ↵
- ↵
- ↵
- ↵
- ↵















